编者按:
新生儿糖尿病(NDM)是一种出生后6个月内起病的极早发糖尿病,其本质并非典型的1型糖尿病,而是以单基因/表观遗传异常驱动的β细胞功能缺陷为主要特征。近年来,随着分子遗传学进展,已有超过40个与NDM相关的致病基因/位点被报道,其中部分基因型已可直接指导治疗选择,使NDM成为精准诊疗的代表性疾病。
在第二十二届北大糖尿病论坛(PUDF 2026)期间,北京协和医院内分泌科肖新华教授围绕“新生儿糖尿病的疾病修正治疗”作专题报告。报告系统阐述了NDM的病因学机制、基因分型路径、疾病修正治疗策略及临床落地实践,旨在为NDM的规范化诊疗提供参考[1]。

一、什么是NDM?
NDM通常定义为出生后6个月内发病的糖尿病,少数病例可延伸至6~12个月(尤其在抗体阴性或单基因表型明确时)。NDM的临床负担远不止于控糖问题。新生儿体重低、进食不稳定、合并症多,严重或反复的高血糖、低血糖、脱水及糖尿病酮症酸中毒(DKA)等代谢紊乱可能增加神经系统发育风险;部分患儿的神经表型还与特定致病基因本身有关[1-5]。
基于临床病程和预后特征,NDM在临床上被分为三类[2](图1):
暂时性新生儿糖尿病(TNDM):约占45%,新生儿期发病后可在婴幼儿期缓解,但青春期或成年后存在复发风险;
永久性新生儿糖尿病(PNDM):约占45%,需终身治疗;
综合征型NDM:约占10%,常累及肝脏、免疫系统、骨骼、外分泌胰腺等多器官,如Wolcott-Rallison综合征(WRS)、IPEX综合征等。
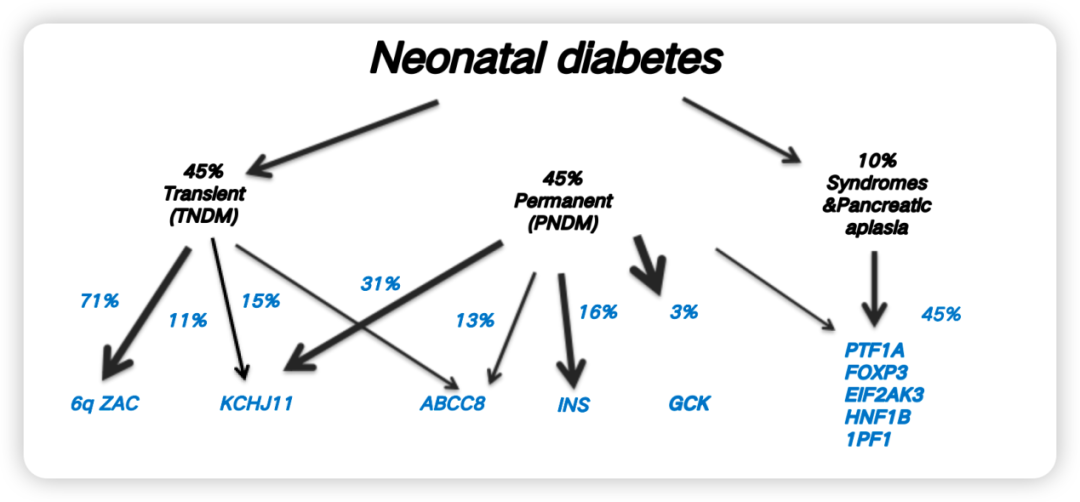
图1. NDM的临床分类
NDM临床分型只是第一步,真正决定治疗方向的,是其背后的病因机制。基于病因,NDM的发病机制可分为五类(图2)[2]:
第一类是β细胞刺激-分泌偶联通路异常:以KCNJ11、ABCC8为代表,核心病理是激活性突变导致KATP通道关闭受阻导致胰岛素分泌障碍;
第二类是胰岛素合成/折叠/内质网应激:涉及INS、EIF2AK3等基因;其中INS突变多与前胰岛素错误折叠及内质网应激相关,EIF2AK3(PERK)缺陷则可造成内质网应激应答障碍并导致β细胞及多器官受累;
第三类是胰腺发育缺陷/β细胞量不足:包括PDX1、PTF1A、GATA6、RFX6等,常伴胰腺发育不全和外分泌功能不全;
第四类是表观遗传/印记异常:以6q24(PLAGL1/HYMAI)为代表,呈现典型的“早发-缓解-复发”自然史;
第五类是免疫调控缺陷:以FOXP3(IPEX综合征)为代表,源于Treg功能缺陷导致多器官自身免疫。
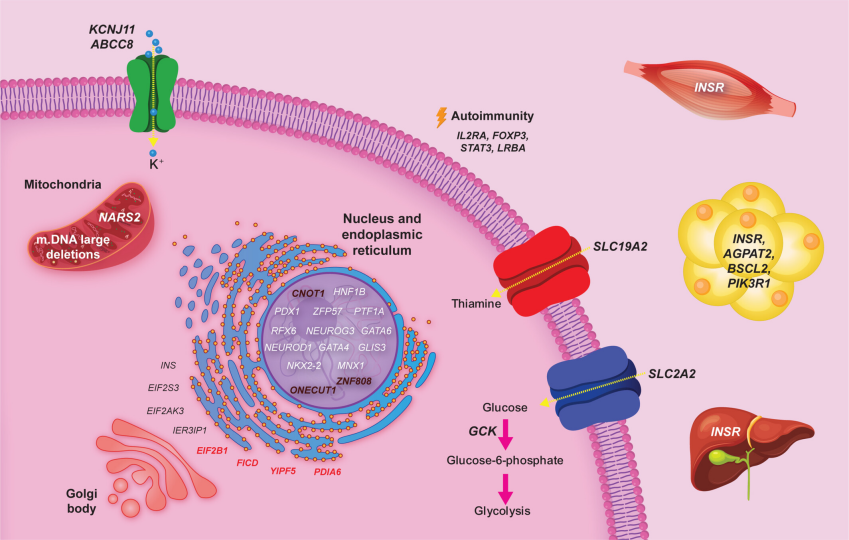
图2. 新生儿糖尿病相关基因
但理解NDM,关键不在记基因,而是在于建立“基因–机制–表型–治疗四联”框架。基因决定病理机制,不同的基因突变对应着不同的分子通路异常;病理机制又决定了表型特征;表型特征反过来提示检测方向;最终的检测结果又会改写治疗路径。以KCNJ11/ABCC8基因为例,其突变导致KATP通道持续开放,引发高血糖及部分患者的神经症状,这一表型线索提示可从胰岛素治疗转换为磺脲类药物(SU),从而实现精准治疗[4-8]。
如此循环往复,形成一个从基因出发、回归临床治疗的正向闭环,这才是NDM精准诊疗的真正内涵。
二、如何识别NDM?从怀疑到遗传检测路径
进行成功的NDM修正治疗,其关键在于早期识别患者,而这依赖临床医生对NDM的高度警惕和系统评估。
1、临床高度怀疑NDM的情形
6至12个月发病且自身抗体阴性或表型高度提示单基因病因;
伴有低出生体重或宫内生长迟缓;
无典型1型糖尿病家族背景或自身抗体模式不支持;
同时合并神经、肝脏、免疫、外分泌胰腺发育异常或畸形线索。
这些线索均提示疾病可能源于单基因突变或印记异常,而非自身免疫破坏。
急性期管理的核心在于及时纠正脱水、酸碱失衡及电解质紊乱,尽快控制高血糖和酮症,维持循环稳定,并尽可能降低高渗、酸中毒及低灌注对脑和其他重要器官的损伤风险。多数患儿需尽早启动胰岛素支持治疗,可根据病情选择静脉胰岛素过渡至皮下注射或胰岛素泵治疗,并在条件允许时结合持续葡萄糖监测(CGM)以减少血糖波动[2]。
3、基础实验室检查与综合征筛查
患儿病情稳定后,应进行相应的实验室检查(图3),不仅用于判断当前代谢状态,还为后续病因分型提供重要线索[2]。

图3. NDM的基础检查
扩大检查范围:对疑似综合征型患儿,建议根据临床线索扩展检查范围,包括肝肾功能、血常规、甲状腺功能、心肾超声,以及必要时的听力、营养和生长发育评估。例如,肝功能异常可提示Wolcott-Rallison综合征,难治性腹泻和湿疹提示IPEX综合征,听力异常和贫血则可提示TRMA综合征。
4、遗传与表观遗传检测
对生后6个月内起病的糖尿病患儿,建议尽快完成遗传学检测。
实践中可采用“NGS基因panel或WES/WGS + 6q24甲基化、拷贝数及UPD评估”的并行检测策略。这一组合既能覆盖常见单基因病因(KCNJ11、ABCC8、INS及多种综合征相关基因),也能识别6q24相关印记异常这一重要机制[1,2,5]。
“快速且全面”的筛查策略优于“逐个猜测”。大型国际队列提示,综合遗传与表观遗传检测策略可在大多数病例中明确病因,且越早检测,越可能在临床表型尚未完全展开前完成诊断,从而提前实施疾病修正治疗、综合征监测和长期随访规划。
三、多层级、精准的NDM疾病修正治疗
NDM的疾病修正治疗并非单一路径,而是基于不同病因机制,形成四种不同层级的干预模式[2](图4)。

图4. NDM的四层干预模式
1、第一层:通道纠正——KATP突变→磺脲类(SU)[4,7,9-11]
KCNJ11或ABCC8突变导致KATP通道持续开放,β细胞无法正常去极化,胰岛素分泌受阻。磺脲类药物可结合SUR1并促进KATP通道关闭,绕过突变通道对ATP关闭信号反应不足的问题,从而恢复β细胞去极化和胰岛素分泌,是通道修正的代表性治疗。
多数明确KCNJ11或ABCC8致病性激活变异的患者可在严密监测下转为磺脲治疗并停用胰岛素,HbA1c通常明显改善;但转换期仍需关注低血糖、胃肠反应及婴幼儿进食波动。现有研究提示,KCNJ11/ABCC8相关NDM患者接受磺脲治疗后,部分神经发育障碍(如肌张力、运动能力、注意力或癫痫相关表现)可能改善,且越早启动治疗,潜在获益越大,但获益程度受基因型和个体差异影响。
这是四种路径中证据最成熟、临床可操作性最强的一级干预。
2、第二层:代谢补充——SLC19A2→硫胺素治疗[2-5,8,12,13]
SLC19A2缺陷导致硫胺素转运异常,临床表现为糖尿病、巨幼红细胞贫血和感音神经性耳聋(TRMA综合征)。
硫胺素治疗可降低胰岛素需求,部分患者可阶段性停用胰岛素;但糖尿病可随年龄进展,听力损害对硫胺素反应通常有限,因此仍需长期随访和多学科管理。
此路径属于“补充缺失底物”的代谢干预,干预方式相对简单,但适用人群较为特定。
3、第三层:免疫重建——FOXP3/IPEX→造血干细胞移植(HSCT)/免疫抑制[2-5,8,12,13]
在IPEX综合征中,糖尿病并非孤立问题,而是全身免疫失衡的一部分。关键临床线索包括难治性腹泻、湿疹、甲状腺或血液系统异常。
治疗策略上,可根据病情以免疫抑制作为桥接;部分患者在多学科评估后可考虑造血干细胞移植,以实现免疫重建。
此路径的干预强度和风险显著高于前两层,但在严格筛选下可改变疾病整体进程。
4、第四层:器官/功能替代——EIF2AK3/WRS→肝移植/多器官移植[2,13]
Wolcott-Rallison综合征的核心风险不仅在于高血糖,更在于反复发作的急性肝衰竭。
胰岛素仍是糖尿病的基础治疗,但在严格筛选下,在极少数严格筛选病例中,肝移植或多器官移植可能作为救治策略并改善预后,但相关证据多来自病例或小队列,需由肝病和移植团队个体化评估。
这是四种路径中干预创伤最大、风险最高的一级,但对特定患儿而言,也是唯一可能改变致死性自然史的选项。
四、个体化治疗算法与临床落地
上述四层干预模式,在临床实践中究竟是如何落地的呢?基因分型结果出来后,医生应该如何一步步实施个体化治疗?这就需要对不同亚型的治疗路径进行具体拆解和规范。
具体而言,在急性期稳定化治疗及完成基因检测之后,NDM治疗路径根据分型结果进入不同的病因导向路径[2]。
1、KATP突变转磺脲实践要点[7,9-11]
KCNJ11或ABCC8相关的新生儿糖尿病是最典型的疾病修正治疗亚型。
转换磺脲治疗的最佳前提是明确KCNJ11或ABCC8致病性激活变异;若仅为高度怀疑,应在有经验的专科中心、严密血糖监测和充分告知风险的前提下谨慎实施。实践中可根据患儿年龄、病情稳定性、家庭管理能力及中心经验,选择住院快速转治或门诊逐步滴定方案。总体原则是在逐步增加格列本脲等磺脲类药物剂量的同时,动态下调胰岛素剂量,避免因转换过快或监测不足带来低血糖或控制不稳。
疗效评估不应仅看是否停用胰岛素,还应关注血糖质量、持续葡萄糖监测时间范围及神经发育结局。
2、特殊亚型管理要点[2,5,13,14]
6q24相关TNDM:初发阶段常需胰岛素支持,随后可逐渐缓解;但缓解不等于治愈,青春期、妊娠或成年后仍有复发风险,需长期监测代谢状态并在高风险阶段加强随访[5]。
IPEX综合征:糖尿病只是全身免疫失衡的一部分,应联合免疫科、移植团队评估免疫抑制桥接和造血干细胞移植时机。
Wolcott-Rallison综合征:核心风险在于反复急性肝衰竭,需联合肝病和移植团队强化肝功能监测,评估肝移植或多器官移植可能。
NDM的机制导向治疗和精准支持治疗取得了重要进展,当前仍存在很多未决问题:磺脲类药物对神经发育获益的因果链仍需更高质量证据;6q24相关TNDM在复发期的最佳口服治疗策略尚未明确;综合征型NDM的最佳干预时机和路径尚未标准化;未明病因NDM提示现有检测能力和机制认知仍存在空白。因此,NDM的精准诊疗还需要更多高质量的循证医学证据。
结语
新生儿糖尿病是精准诊疗的代表性疾病。基因结果可直接改写治疗方式——从胰岛素替代转向磺脲口服、硫胺素补充、免疫重建或器官替代。当前最重要的临床启示是:生后6个月内发病的糖尿病,应优先考虑单基因或印记异常;快速遗传与表观遗传分型,不是附加项,而是决定后续治疗路径的基础设施。越早完成分型,越可能争取更大的长期获益。
未来,NDM管理将从“精准诊断”进一步走向“精准修正”,为每一例患儿设计专属的病因导向治疗路径。
参考文献
1.Glaser N, et al. Pediatr Diabetes. 2022. 23(7): 835-856.
2.Beltrand J, et al. Front Pediatr. 2020. 8: 540718.
3.De Franco E, et al. Lancet. 2015. 386(9997): 957-963.
4.De Leon DD, et al. GeneReviews: Permanent Neonatal Diabetes Mellitus.
5.Temple IK, et al. GeneReviews: Diabetes Mellitus, 6q24-Related Transient Neonatal.
6.Barbetti F, et al. J Diabetes Investig. 2024. 15(12): 1711-1724.
7.Pearson ER, et al. N Engl J Med. 2006. 355(5): 467-477.
8.Ben-Skowronek I, et al. Genes (Basel). 2021. 12(3):323.
9.Beltrand J, et al. Diabetes Care. 2015. 38(11):2033-2041.
10.Bowman P, et al. Lancet Diabetes Endocrinol. 2018. 6(8):637-646.
11.Bowman P, et al. Diabetes Care. 2021. 44(1):35-42.
12. Habeb AM, et al. Diabetologia. 2018. 61(5):1027-1036.
13. Aldrian D, et al. Liver Int. 2024. 44(3):811-822.
14. Hou AN, et al. J Clin Immunol. 2023. 43(5):979-988. 2 comments
新生儿糖尿病(NDM)是一种出生后6个月内起病的极早发糖尿病,其本质并非典型的1型糖尿病,而是以单基因/表观遗传异常驱动的β细胞功能缺陷为主要特征。近年来,随着分子遗传学进展,已有超过40个与NDM相关的致病基因/位点被报道,其中部分基因型已可直接指导治疗选择,使NDM成为精准诊疗的代表性疾病。
在第二十二届北大糖尿病论坛(PUDF 2026)期间,北京协和医院内分泌科肖新华教授围绕“新生儿糖尿病的疾病修正治疗”作专题报告。报告系统阐述了NDM的病因学机制、基因分型路径、疾病修正治疗策略及临床落地实践,旨在为NDM的规范化诊疗提供参考[1]。
一、什么是NDM?
NDM通常定义为出生后6个月内发病的糖尿病,少数病例可延伸至6~12个月(尤其在抗体阴性或单基因表型明确时)。NDM的临床负担远不止于控糖问题。新生儿体重低、进食不稳定、合并症多,严重或反复的高血糖、低血糖、脱水及糖尿病酮症酸中毒(DKA)等代谢紊乱可能增加神经系统发育风险;部分患儿的神经表型还与特定致病基因本身有关[1-5]。
基于临床病程和预后特征,NDM在临床上被分为三类[2](图1):
暂时性新生儿糖尿病(TNDM):约占45%,新生儿期发病后可在婴幼儿期缓解,但青春期或成年后存在复发风险;
永久性新生儿糖尿病(PNDM):约占45%,需终身治疗;
综合征型NDM:约占10%,常累及肝脏、免疫系统、骨骼、外分泌胰腺等多器官,如Wolcott-Rallison综合征(WRS)、IPEX综合征等。
图1. NDM的临床分类
NDM临床分型只是第一步,真正决定治疗方向的,是其背后的病因机制。基于病因,NDM的发病机制可分为五类(图2)[2]:
第一类是β细胞刺激-分泌偶联通路异常:以KCNJ11、ABCC8为代表,核心病理是激活性突变导致KATP通道关闭受阻导致胰岛素分泌障碍;
第二类是胰岛素合成/折叠/内质网应激:涉及INS、EIF2AK3等基因;其中INS突变多与前胰岛素错误折叠及内质网应激相关,EIF2AK3(PERK)缺陷则可造成内质网应激应答障碍并导致β细胞及多器官受累;
第三类是胰腺发育缺陷/β细胞量不足:包括PDX1、PTF1A、GATA6、RFX6等,常伴胰腺发育不全和外分泌功能不全;
第四类是表观遗传/印记异常:以6q24(PLAGL1/HYMAI)为代表,呈现典型的“早发-缓解-复发”自然史;
第五类是免疫调控缺陷:以FOXP3(IPEX综合征)为代表,源于Treg功能缺陷导致多器官自身免疫。
图2. 新生儿糖尿病相关基因
但理解NDM,关键不在记基因,而是在于建立“基因–机制–表型–治疗四联”框架。基因决定病理机制,不同的基因突变对应着不同的分子通路异常;病理机制又决定了表型特征;表型特征反过来提示检测方向;最终的检测结果又会改写治疗路径。以KCNJ11/ABCC8基因为例,其突变导致KATP通道持续开放,引发高血糖及部分患者的神经症状,这一表型线索提示可从胰岛素治疗转换为磺脲类药物(SU),从而实现精准治疗[4-8]。
如此循环往复,形成一个从基因出发、回归临床治疗的正向闭环,这才是NDM精准诊疗的真正内涵。
二、如何识别NDM?从怀疑到遗传检测路径
进行成功的NDM修正治疗,其关键在于早期识别患者,而这依赖临床医生对NDM的高度警惕和系统评估。
1、临床高度怀疑NDM的情形
当出现以下情形时,临床应优先按照NDM诊断路径进行评估,而非简单归类为1型糖尿病[1-4]:
6至12个月发病且自身抗体阴性或表型高度提示单基因病因;
伴有低出生体重或宫内生长迟缓;
无典型1型糖尿病家族背景或自身抗体模式不支持;
同时合并神经、肝脏、免疫、外分泌胰腺发育异常或畸形线索。
这些线索均提示疾病可能源于单基因突变或印记异常,而非自身免疫破坏。
2、急性期评估与管理
急性期管理的核心在于及时纠正脱水、酸碱失衡及电解质紊乱,尽快控制高血糖和酮症,维持循环稳定,并尽可能降低高渗、酸中毒及低灌注对脑和其他重要器官的损伤风险。多数患儿需尽早启动胰岛素支持治疗,可根据病情选择静脉胰岛素过渡至皮下注射或胰岛素泵治疗,并在条件允许时结合持续葡萄糖监测(CGM)以减少血糖波动[2]。
3、基础实验室检查与综合征筛查
患儿病情稳定后,应进行相应的实验室检查(图3),不仅用于判断当前代谢状态,还为后续病因分型提供重要线索[2]。
图3. NDM的基础检查
胰腺超声:关注是否存在胰腺发育不全或缺如,这对提示胰腺发育相关基因异常具有重要价值,同时也有助于预判外分泌胰腺功能不全风险。若表型提示综合征型NDM,还应结合心脏、肾脏等影像学检查,进一步识别多系统受累。
C肽和胰岛素水平:以评估内源性胰岛β细胞分泌功能,同时进行自身抗体检测(GAD、IA-2、ZnT8,及IAA)。但即使抗体阳性,生后6个月内起病者仍不能据此排除单基因病因;遗传检测不应因抗体结果而延后。扩大检查范围:对疑似综合征型患儿,建议根据临床线索扩展检查范围,包括肝肾功能、血常规、甲状腺功能、心肾超声,以及必要时的听力、营养和生长发育评估。例如,肝功能异常可提示Wolcott-Rallison综合征,难治性腹泻和湿疹提示IPEX综合征,听力异常和贫血则可提示TRMA综合征。
4、遗传与表观遗传检测
对生后6个月内起病的糖尿病患儿,建议尽快完成遗传学检测。
实践中可采用“NGS基因panel或WES/WGS + 6q24甲基化、拷贝数及UPD评估”的并行检测策略。这一组合既能覆盖常见单基因病因(KCNJ11、ABCC8、INS及多种综合征相关基因),也能识别6q24相关印记异常这一重要机制[1,2,5]。
“快速且全面”的筛查策略优于“逐个猜测”。大型国际队列提示,综合遗传与表观遗传检测策略可在大多数病例中明确病因,且越早检测,越可能在临床表型尚未完全展开前完成诊断,从而提前实施疾病修正治疗、综合征监测和长期随访规划。
三、多层级、精准的NDM疾病修正治疗
NDM的疾病修正治疗并非单一路径,而是基于不同病因机制,形成四种不同层级的干预模式[2](图4)。
图4. NDM的四层干预模式
1、第一层:通道纠正——KATP突变→磺脲类(SU)[4,7,9-11]
KCNJ11或ABCC8突变导致KATP通道持续开放,β细胞无法正常去极化,胰岛素分泌受阻。磺脲类药物可结合SUR1并促进KATP通道关闭,绕过突变通道对ATP关闭信号反应不足的问题,从而恢复β细胞去极化和胰岛素分泌,是通道修正的代表性治疗。
多数明确KCNJ11或ABCC8致病性激活变异的患者可在严密监测下转为磺脲治疗并停用胰岛素,HbA1c通常明显改善;但转换期仍需关注低血糖、胃肠反应及婴幼儿进食波动。现有研究提示,KCNJ11/ABCC8相关NDM患者接受磺脲治疗后,部分神经发育障碍(如肌张力、运动能力、注意力或癫痫相关表现)可能改善,且越早启动治疗,潜在获益越大,但获益程度受基因型和个体差异影响。
这是四种路径中证据最成熟、临床可操作性最强的一级干预。
2、第二层:代谢补充——SLC19A2→硫胺素治疗[2-5,8,12,13]
SLC19A2缺陷导致硫胺素转运异常,临床表现为糖尿病、巨幼红细胞贫血和感音神经性耳聋(TRMA综合征)。
硫胺素治疗可降低胰岛素需求,部分患者可阶段性停用胰岛素;但糖尿病可随年龄进展,听力损害对硫胺素反应通常有限,因此仍需长期随访和多学科管理。
此路径属于“补充缺失底物”的代谢干预,干预方式相对简单,但适用人群较为特定。
3、第三层:免疫重建——FOXP3/IPEX→造血干细胞移植(HSCT)/免疫抑制[2-5,8,12,13]
在IPEX综合征中,糖尿病并非孤立问题,而是全身免疫失衡的一部分。关键临床线索包括难治性腹泻、湿疹、甲状腺或血液系统异常。
治疗策略上,可根据病情以免疫抑制作为桥接;部分患者在多学科评估后可考虑造血干细胞移植,以实现免疫重建。
此路径的干预强度和风险显著高于前两层,但在严格筛选下可改变疾病整体进程。
4、第四层:器官/功能替代——EIF2AK3/WRS→肝移植/多器官移植[2,13]
Wolcott-Rallison综合征的核心风险不仅在于高血糖,更在于反复发作的急性肝衰竭。
胰岛素仍是糖尿病的基础治疗,但在严格筛选下,在极少数严格筛选病例中,肝移植或多器官移植可能作为救治策略并改善预后,但相关证据多来自病例或小队列,需由肝病和移植团队个体化评估。
这是四种路径中干预创伤最大、风险最高的一级,但对特定患儿而言,也是唯一可能改变致死性自然史的选项。
四、个体化治疗算法与临床落地
上述四层干预模式,在临床实践中究竟是如何落地的呢?基因分型结果出来后,医生应该如何一步步实施个体化治疗?这就需要对不同亚型的治疗路径进行具体拆解和规范。
具体而言,在急性期稳定化治疗及完成基因检测之后,NDM治疗路径根据分型结果进入不同的病因导向路径[2]。
1、KATP突变转磺脲实践要点[7,9-11]
KCNJ11或ABCC8相关的新生儿糖尿病是最典型的疾病修正治疗亚型。
转换磺脲治疗的最佳前提是明确KCNJ11或ABCC8致病性激活变异;若仅为高度怀疑,应在有经验的专科中心、严密血糖监测和充分告知风险的前提下谨慎实施。实践中可根据患儿年龄、病情稳定性、家庭管理能力及中心经验,选择住院快速转治或门诊逐步滴定方案。总体原则是在逐步增加格列本脲等磺脲类药物剂量的同时,动态下调胰岛素剂量,避免因转换过快或监测不足带来低血糖或控制不稳。
疗效评估不应仅看是否停用胰岛素,还应关注血糖质量、持续葡萄糖监测时间范围及神经发育结局。
2、特殊亚型管理要点[2,5,13,14]
6q24相关TNDM:初发阶段常需胰岛素支持,随后可逐渐缓解;但缓解不等于治愈,青春期、妊娠或成年后仍有复发风险,需长期监测代谢状态并在高风险阶段加强随访[5]。
IPEX综合征:糖尿病只是全身免疫失衡的一部分,应联合免疫科、移植团队评估免疫抑制桥接和造血干细胞移植时机。
Wolcott-Rallison综合征:核心风险在于反复急性肝衰竭,需联合肝病和移植团队强化肝功能监测,评估肝移植或多器官移植可能。
NDM的机制导向治疗和精准支持治疗取得了重要进展,当前仍存在很多未决问题:磺脲类药物对神经发育获益的因果链仍需更高质量证据;6q24相关TNDM在复发期的最佳口服治疗策略尚未明确;综合征型NDM的最佳干预时机和路径尚未标准化;未明病因NDM提示现有检测能力和机制认知仍存在空白。因此,NDM的精准诊疗还需要更多高质量的循证医学证据。
结语
新生儿糖尿病是精准诊疗的代表性疾病。基因结果可直接改写治疗方式——从胰岛素替代转向磺脲口服、硫胺素补充、免疫重建或器官替代。当前最重要的临床启示是:生后6个月内发病的糖尿病,应优先考虑单基因或印记异常;快速遗传与表观遗传分型,不是附加项,而是决定后续治疗路径的基础设施。越早完成分型,越可能争取更大的长期获益。
未来,NDM管理将从“精准诊断”进一步走向“精准修正”,为每一例患儿设计专属的病因导向治疗路径。
参考文献
1.Glaser N, et al. Pediatr Diabetes. 2022. 23(7): 835-856.
2.Beltrand J, et al. Front Pediatr. 2020. 8: 540718.
3.De Franco E, et al. Lancet. 2015. 386(9997): 957-963.
4.De Leon DD, et al. GeneReviews: Permanent Neonatal Diabetes Mellitus.
5.Temple IK, et al. GeneReviews: Diabetes Mellitus, 6q24-Related Transient Neonatal.
6.Barbetti F, et al. J Diabetes Investig. 2024. 15(12): 1711-1724.
7.Pearson ER, et al. N Engl J Med. 2006. 355(5): 467-477.
8.Ben-Skowronek I, et al. Genes (Basel). 2021. 12(3):323.
9.Beltrand J, et al. Diabetes Care. 2015. 38(11):2033-2041.
10.Bowman P, et al. Lancet Diabetes Endocrinol. 2018. 6(8):637-646.
11.Bowman P, et al. Diabetes Care. 2021. 44(1):35-42.
12. Habeb AM, et al. Diabetologia. 2018. 61(5):1027-1036.
13. Aldrian D, et al. Liver Int. 2024. 44(3):811-822.
14. Hou AN, et al. J Clin Immunol. 2023. 43(5):979-988. 2 comments










 京公网安备 11010502033361号
京公网安备 11010502033361号
发布留言