编者按
遗传性胰岛素抵抗综合征为胰岛素受体及下游通路基因缺陷诱发的罕见内分泌疾病,多合并生长发育、生殖系统多器官受累,易误诊为1型糖尿病、多囊卵巢综合征。SHORT综合征由PIK3R1基因杂合突变致病,突变破坏PI3K调节亚基功能,阻滞INSR-AKT葡萄糖代谢信号传导,形成空腹平稳、碳水摄入后餐后血糖骤升的特征性糖代谢模式。该病典型表现包含宫内发育迟缓、特殊面容、出牙延迟、矮小、卵巢囊肿、高雄激素血症,糖代谢异常发生率超60%。患者常对大剂量胰岛素反应差,需依靠饮食调控、个体化口服药控制血糖,确诊依赖葡萄糖钳夹试验与基因测序,需与Donohue等重度胰岛素抵抗疾病鉴别。本病临床少见,诊疗方案缺乏统一规范。本文中,广东医科大学附属医院骆梓恒医生分享1例以糖尿病酮症起病、合并多系统畸形的青少年女性SHORT综合征病例,完整分析其临床特点与个体化治疗思路。
患者基本信息
患者女性,16岁。因“发现血糖升高1年半”入院。患者1年半前因上呼吸道感染于当地医院测随机末梢血糖18.4mmol/L,尿常规示尿糖(++)、尿酮体(+++),血气分析未见酸中毒,HbA1c 12.1%,诊断为“糖尿病酮症”,给予补液、胰岛素泵降糖等对症处理后尿酮体消失。出院后予门冬胰岛素16U三餐前、甘精胰岛素20U睡前皮下注射,二甲双胍缓释片0.5g 2次/日、消渴冲剂1袋3次/日治疗,监测空腹血糖7~10mmol/L,餐后2小时血糖8~16mmol/L,无低血糖发生。患者后续自行停用胰岛素及二甲双胍,仅口服消渴冲剂治疗,监测空腹血糖8~13mmol/L,餐后2小时血糖15~18mmol/L,加强运动后空腹血糖可降至4~5mmol/L,但进食后餐后2小时血糖明显升高。患者系第1胎第1产,足月剖宫产,出生时无难产及窒息;出生体重2.4kg(-2SD),身长42cm(-2SD)。坐立、爬行、站立、智力发育与同龄儿童相仿;1岁2个月萌芽,牙齿排列不齐。自幼身高较同龄儿童矮,10岁时因身高偏矮测IGF-1未见异常,近2年身高增长2cm。第二性征发育时间不详,至今无月经来潮。父母非近亲婚配,父亲身高170cm,母亲身高156cm,无糖尿病家族史。
11岁发现右侧卵巢囊肿,行手术治疗;12岁时再次发现左侧卵巢囊肿行手术治疗。
体格检查
血压110/74mmHg,身高140cm(-2SD),上部量72cm,下部量68cm,指尖距130.5cm,体重31.5kg,BMI 16.1kg/m2。体型消瘦,全身皮肤偏黑、粗糙、四肢多毛;额头膨隆,眼距增宽,眼窝深陷,嘴角下垂,下颌尖且短,双耳大且耳位低;全身皮下脂肪少,颈部呈黑棘皮样改变,双足短指畸形,无明显肘外翻。双侧乳房Tanner Ⅲ期,外阴发育无异常,阴毛Tanner Ⅳ期,腋毛稀疏。
辅助检查
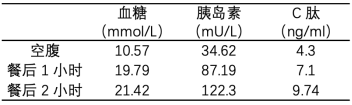
血常规、肝肾功能、血脂、促肾上腺皮质激素、皮质醇、甲状腺功能、泌乳素均未见异常。尿常规示尿糖1000mg/dl,尿酮体阴性、尿蛋白阴性;HbA1c 11.4%;正常餐试验示血糖及胰岛素明显升高(表1),高胰岛素正常葡萄糖钳夹试验示葡萄糖输注速率GIR值为4.6mg/(min·kg)(中度胰岛素抵抗);谷氨酸脱羧酶自身抗体、胰岛素抗体、胰岛细胞抗体均阴性;IGF-1 617ng/ml;睾酮2.85nmol/L,黄体生成素16.43mIU/ml。骨龄15岁11个月,骨骺闭合。妇科超声:子宫大小约2.9cm×1.3cm×2.0cm,内膜厚约0.4cm,宫腔线显示尚清晰;左卵巢大小约3.6cm×1.8cm×1.9cm,内超过4mm的卵泡有5个,大者直径约0.4cm,右卵巢大小3.0cm×1.5cm×1.8cm,内超过4mm的卵泡有6个,大者直径约0.6cm,双附件区未见异常回声。听力检测正常。眼科检查:右眼裸眼视力0.3,左眼裸眼视力0.4,双眼矫正视力1.0(自10岁起戴近视眼镜);右眼眼压23mmHg;左眼眼压20mmHg;眼底未见异常。全外显子测序:患儿PIK3R1基因第14外显子c.1945C>T杂合突变,致使第649位氨基酸由精氨酸变为色氨酸(p.Arg649Trp)。Sanger测序家系验证显示其父母均未携带此突变,提示为新生突变。
表1. 患者正常餐试验结果
诊治经过
结合患者症状、体征、辅助检查,入院诊断考虑为:SHORT综合征。
入院后给予胰岛素降糖,胰岛素用量高达94U/天(3U/kg)时空腹血糖波动在9~14mmol/L,餐后2小时血糖波动在18~22mmol/L;禁食及生酮饮食后患者血糖波动在5.4~10mmol/L,但进食碳水化合物后餐后2小时血糖可升高至23.7mmol/L;正常饮食情况下给予二甲双胍0.5g 3次/日治疗,空腹血糖波动在6.3~14mmol/L,餐后2小时血糖波动在13~16mmol/L,加用阿卡波糖50mg 3次/日后空腹血糖波动在5.9~7.3mmol/L,餐后2小时波动在7~10mmol/L。因患者骨骺闭合,未使用生长激素治疗。患者至今无月经来潮且有高雄激素血症,虽妇科超声未达到多囊卵巢诊断标准,因患者处于青春期,仍考虑患者存在多囊卵巢综合征,患者口服二甲双胍3个月后仍无月经来潮。
病例诊疗思考与总结
SHORT以特征性的身材矮小、关节过度伸展和(或)腹股沟疝、眼球凹陷、Rieger异常、萌芽延迟为临床特征。SHORT综合征的其他特征包括宫内发育迟缓、特殊面容(三角脸、前额突出、眼球深陷、耳位低伴大耳、嘴角下弯、小下颌)、脂肪萎缩、胰岛素抵抗或糖尿病,语言延迟、感音神经性耳聋临床相对少见。遗传性胰岛素抵抗综合征中的Donohue综合征Rabson-Mendenhall综合征是罕见的常染色体隐性遗传病,位于人类第19号常染色体短臂(19p13.2)上的胰岛素受体(Insulin Receptor,INSR)基因突变所致,临床亦可表现为宫内发育迟缓、眼距宽、扁平鼻、耳位低、多毛、皮下脂肪萎缩、胰岛素抵抗及糖尿病(空腹低血糖伴极度胰岛素抵抗),易与SHORT综合征混淆,但后者常表现为极度胰岛素抵抗[空腹胰岛素>70mU/L,餐后2小时胰岛素>350mU/L,GIR>2mg/(min·kg)],基因检测利于两者鉴别[1]。
本例患者以糖尿病来诊,大剂量胰岛素治疗效果不佳,且发病年龄小、体型消瘦、矮身材、眼球凹陷、萌芽延迟、面部特征、卵巢囊肿,提示存在遗传性疾病导致的胰岛素抵抗;入院后查胰岛素水平仅轻度升高,GIR提示中度胰岛素抵抗,提示SHORT综合征可能性大,基因检测证实为SHORT综合征。
SHORT综合征主要是由于人类第5号染色体的PIK3R1基因突变引起,该基因是SHORT综合征的唯一致病基因。PIK3R1基因含有15个内含子和16个外显子,通过编码磷脂酰肌醇-3激酶(Phosphatidylin-ositol-3-kinase,PI3K)的调节亚基(p85α、p55α和p50α),参与多个信号通路传导,包括生长激素受体(Growth hormone receptor,GHR)、胰岛素样生长因子1受体(Insulin-like growth factor-1 receptor,IGF-1R)、INSR/胰岛素受体底物-1(Insulin receptor substrate-1,IRS-1)以及免疫相关的信号通路。通过激活AKT/mTOR途径,调节脂肪分化、细胞生长和增殖、胰岛素信号传导等。PI3K的调节亚基含有SH2和SH3结构域,其中SH2结构域在PI3K的调剂活性中起关键作用,目前已报道的10个PIK3R1基因突变均位于该结构域。PIK3R1基因第14外显子c.1945C>T突变是热点突变,常引起典型临床症状,本例患者也是该位点的杂合突变。PIK3R1基因的突变类型和临床表型之间的相关性目前尚不明确,c.1106C>T突变表现为三角脸及正常身高,c.1945C>T、c.1906/1907C>T和c.1971T>G基因突变患者表现为Rieger异常[2,3]。
INSR-IRS-1-PI3K-AKT信号通路通过调节外周胰岛素靶组织葡萄糖转运蛋白-4(Glucose transporter-4,GLUT-4)和GLUT-2的表达及转位介导葡萄糖摄取,该信号通路的任何基因突变均可导致胰岛素抵抗及糖尿病的发生。PIK3R1基因突变导致的PI3K调节亚基p85α功能缺失使其不能与催化亚基p110结合,致使INSR信号通路受阻导致胰岛素抵抗及糖尿病。本例患者表现为空腹正常或偏低血糖,餐后血糖异常升高,这种昼夜节律的血糖模式也见于INSR杂合突变导致的胰岛素抵抗患者,这是INSR及受体后信号通路障碍导致胰岛素抵抗患者的血糖谱特点,少食多餐或低碳水高蛋白质饮食可改善此种血糖的昼夜节律波动。该患者正常饮食或高碳水化合物饮食时血糖明显升高,但生酮饮食及禁食情况下血糖可维持在正常范围,符合PIK3R1基因突变致SHORT综合征糖尿病的血糖谱特点。二甲双胍是治疗SHORT综合征胰岛素抵抗及糖尿病的关键药物,但也有报道二甲双胍有加重胰岛素抵抗的可能。在二甲双胍单药治疗疗效差时,加用钠-葡萄糖协同转运蛋白2抑制剂(SGLT2i)也可获得良好疗效,SGLT2i不仅可通过降低肾糖阈、增加尿糖排泄量来降低日常食物摄入后导致的高血糖,而且还可以缓解轻微的夜间低血糖,但存在患者体重进一步减轻的风险[4,5]。本例患者使用二甲双胍治疗后空腹血糖达标但餐后血糖仍较高,结合患者高碳水化合物饮食后餐后血糖异常升高的血糖谱特点,同时避免使用SGLT2i使用后引起的体重进一步下降,给予患者二甲双胍联合阿卡波糖治疗,患者餐后血糖平稳达标。本例患者的治疗方案为SHORT综合征糖尿病的治疗提供了新方案。
SHORT综合征伴有身材矮小,男性成人身高为153.7~167cm,女性身高为141~167cm,生长激素治疗是增加终身高的有效方法,但会导致胰岛素抵抗的风险增加,尤其是合并糖尿病的患者会导致血糖难以控制,对于SHORT综合征患者,应谨慎评估生长激素治疗的益处和风险。本例患者已合并糖尿病,且骨骺已闭合,未再使用生长激素治疗。SHORT综合征亦会因胰岛素抵抗导致PCOS,本例患者虽妇科超声未达到PCOS诊断标准,但第二性征发育好,骨骺已闭合,至今无月经来潮,实验室检查提示睾酮及LH升高,仍考虑存在PCOS,给予二甲双胍治疗3个月后随诊,如仍无月经来潮可加用噻唑烷二酮类药物治疗。
参考文献:
[1] Ogawa W, Araki E, Ishigaki Y, et al. New classification and diagnostic criteria for insulin resistance syndrome.Endocr J. 2022;69(2):107-113.
[2] Shvalb NF. SHORT Syndrome: an Update on Pathogenesis and Clinical Spectrum.Curr Diab Rep. 2022;22(12):571-577.
[3] Innes AM, Neufeld S, Dyment DA.PIK3R1-Related SHORT Syndrome. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds.GeneReviews®. Seattle (WA): University of Washington, Seattle; May 15, 2014.
[4] Masunaga Y, Fujisawa Y, Muramatsu M, et al. Insulin resistant diabetes mellitus in SHORT syndrome: case report and literature review.Endocr J. 2021;68(1):111-117.
[5] Yang Y, Tan SHC, Lim SC, Loh WJ. Diabetes mellitus in SHORT syndrome managed with multi-agent oral therapies: a case report and literature review.Ther Adv Endocrinol Metab. 2025;16:20420188251405363. 2 comments
遗传性胰岛素抵抗综合征为胰岛素受体及下游通路基因缺陷诱发的罕见内分泌疾病,多合并生长发育、生殖系统多器官受累,易误诊为1型糖尿病、多囊卵巢综合征。SHORT综合征由PIK3R1基因杂合突变致病,突变破坏PI3K调节亚基功能,阻滞INSR-AKT葡萄糖代谢信号传导,形成空腹平稳、碳水摄入后餐后血糖骤升的特征性糖代谢模式。该病典型表现包含宫内发育迟缓、特殊面容、出牙延迟、矮小、卵巢囊肿、高雄激素血症,糖代谢异常发生率超60%。患者常对大剂量胰岛素反应差,需依靠饮食调控、个体化口服药控制血糖,确诊依赖葡萄糖钳夹试验与基因测序,需与Donohue等重度胰岛素抵抗疾病鉴别。本病临床少见,诊疗方案缺乏统一规范。本文中,广东医科大学附属医院骆梓恒医生分享1例以糖尿病酮症起病、合并多系统畸形的青少年女性SHORT综合征病例,完整分析其临床特点与个体化治疗思路。
患者基本信息
患者女性,16岁。因“发现血糖升高1年半”入院。患者1年半前因上呼吸道感染于当地医院测随机末梢血糖18.4mmol/L,尿常规示尿糖(++)、尿酮体(+++),血气分析未见酸中毒,HbA1c 12.1%,诊断为“糖尿病酮症”,给予补液、胰岛素泵降糖等对症处理后尿酮体消失。出院后予门冬胰岛素16U三餐前、甘精胰岛素20U睡前皮下注射,二甲双胍缓释片0.5g 2次/日、消渴冲剂1袋3次/日治疗,监测空腹血糖7~10mmol/L,餐后2小时血糖8~16mmol/L,无低血糖发生。患者后续自行停用胰岛素及二甲双胍,仅口服消渴冲剂治疗,监测空腹血糖8~13mmol/L,餐后2小时血糖15~18mmol/L,加强运动后空腹血糖可降至4~5mmol/L,但进食后餐后2小时血糖明显升高。患者系第1胎第1产,足月剖宫产,出生时无难产及窒息;出生体重2.4kg(-2SD),身长42cm(-2SD)。坐立、爬行、站立、智力发育与同龄儿童相仿;1岁2个月萌芽,牙齿排列不齐。自幼身高较同龄儿童矮,10岁时因身高偏矮测IGF-1未见异常,近2年身高增长2cm。第二性征发育时间不详,至今无月经来潮。父母非近亲婚配,父亲身高170cm,母亲身高156cm,无糖尿病家族史。
既往史
11岁发现右侧卵巢囊肿,行手术治疗;12岁时再次发现左侧卵巢囊肿行手术治疗。
体格检查
血压110/74mmHg,身高140cm(-2SD),上部量72cm,下部量68cm,指尖距130.5cm,体重31.5kg,BMI 16.1kg/m2。体型消瘦,全身皮肤偏黑、粗糙、四肢多毛;额头膨隆,眼距增宽,眼窝深陷,嘴角下垂,下颌尖且短,双耳大且耳位低;全身皮下脂肪少,颈部呈黑棘皮样改变,双足短指畸形,无明显肘外翻。双侧乳房Tanner Ⅲ期,外阴发育无异常,阴毛Tanner Ⅳ期,腋毛稀疏。
辅助检查
血常规、肝肾功能、血脂、促肾上腺皮质激素、皮质醇、甲状腺功能、泌乳素均未见异常。尿常规示尿糖1000mg/dl,尿酮体阴性、尿蛋白阴性;HbA1c 11.4%;正常餐试验示血糖及胰岛素明显升高(表1),高胰岛素正常葡萄糖钳夹试验示葡萄糖输注速率GIR值为4.6mg/(min·kg)(中度胰岛素抵抗);谷氨酸脱羧酶自身抗体、胰岛素抗体、胰岛细胞抗体均阴性;IGF-1 617ng/ml;睾酮2.85nmol/L,黄体生成素16.43mIU/ml。骨龄15岁11个月,骨骺闭合。妇科超声:子宫大小约2.9cm×1.3cm×2.0cm,内膜厚约0.4cm,宫腔线显示尚清晰;左卵巢大小约3.6cm×1.8cm×1.9cm,内超过4mm的卵泡有5个,大者直径约0.4cm,右卵巢大小3.0cm×1.5cm×1.8cm,内超过4mm的卵泡有6个,大者直径约0.6cm,双附件区未见异常回声。听力检测正常。眼科检查:右眼裸眼视力0.3,左眼裸眼视力0.4,双眼矫正视力1.0(自10岁起戴近视眼镜);右眼眼压23mmHg;左眼眼压20mmHg;眼底未见异常。全外显子测序:患儿PIK3R1基因第14外显子c.1945C>T杂合突变,致使第649位氨基酸由精氨酸变为色氨酸(p.Arg649Trp)。Sanger测序家系验证显示其父母均未携带此突变,提示为新生突变。
表1. 患者正常餐试验结果
诊治经过
结合患者症状、体征、辅助检查,入院诊断考虑为:SHORT综合征。
入院后给予胰岛素降糖,胰岛素用量高达94U/天(3U/kg)时空腹血糖波动在9~14mmol/L,餐后2小时血糖波动在18~22mmol/L;禁食及生酮饮食后患者血糖波动在5.4~10mmol/L,但进食碳水化合物后餐后2小时血糖可升高至23.7mmol/L;正常饮食情况下给予二甲双胍0.5g 3次/日治疗,空腹血糖波动在6.3~14mmol/L,餐后2小时血糖波动在13~16mmol/L,加用阿卡波糖50mg 3次/日后空腹血糖波动在5.9~7.3mmol/L,餐后2小时波动在7~10mmol/L。因患者骨骺闭合,未使用生长激素治疗。患者至今无月经来潮且有高雄激素血症,虽妇科超声未达到多囊卵巢诊断标准,因患者处于青春期,仍考虑患者存在多囊卵巢综合征,患者口服二甲双胍3个月后仍无月经来潮。
病例诊疗思考与总结
SHORT以特征性的身材矮小、关节过度伸展和(或)腹股沟疝、眼球凹陷、Rieger异常、萌芽延迟为临床特征。SHORT综合征的其他特征包括宫内发育迟缓、特殊面容(三角脸、前额突出、眼球深陷、耳位低伴大耳、嘴角下弯、小下颌)、脂肪萎缩、胰岛素抵抗或糖尿病,语言延迟、感音神经性耳聋临床相对少见。遗传性胰岛素抵抗综合征中的Donohue综合征Rabson-Mendenhall综合征是罕见的常染色体隐性遗传病,位于人类第19号常染色体短臂(19p13.2)上的胰岛素受体(Insulin Receptor,INSR)基因突变所致,临床亦可表现为宫内发育迟缓、眼距宽、扁平鼻、耳位低、多毛、皮下脂肪萎缩、胰岛素抵抗及糖尿病(空腹低血糖伴极度胰岛素抵抗),易与SHORT综合征混淆,但后者常表现为极度胰岛素抵抗[空腹胰岛素>70mU/L,餐后2小时胰岛素>350mU/L,GIR>2mg/(min·kg)],基因检测利于两者鉴别[1]。
本例患者以糖尿病来诊,大剂量胰岛素治疗效果不佳,且发病年龄小、体型消瘦、矮身材、眼球凹陷、萌芽延迟、面部特征、卵巢囊肿,提示存在遗传性疾病导致的胰岛素抵抗;入院后查胰岛素水平仅轻度升高,GIR提示中度胰岛素抵抗,提示SHORT综合征可能性大,基因检测证实为SHORT综合征。
SHORT综合征主要是由于人类第5号染色体的PIK3R1基因突变引起,该基因是SHORT综合征的唯一致病基因。PIK3R1基因含有15个内含子和16个外显子,通过编码磷脂酰肌醇-3激酶(Phosphatidylin-ositol-3-kinase,PI3K)的调节亚基(p85α、p55α和p50α),参与多个信号通路传导,包括生长激素受体(Growth hormone receptor,GHR)、胰岛素样生长因子1受体(Insulin-like growth factor-1 receptor,IGF-1R)、INSR/胰岛素受体底物-1(Insulin receptor substrate-1,IRS-1)以及免疫相关的信号通路。通过激活AKT/mTOR途径,调节脂肪分化、细胞生长和增殖、胰岛素信号传导等。PI3K的调节亚基含有SH2和SH3结构域,其中SH2结构域在PI3K的调剂活性中起关键作用,目前已报道的10个PIK3R1基因突变均位于该结构域。PIK3R1基因第14外显子c.1945C>T突变是热点突变,常引起典型临床症状,本例患者也是该位点的杂合突变。PIK3R1基因的突变类型和临床表型之间的相关性目前尚不明确,c.1106C>T突变表现为三角脸及正常身高,c.1945C>T、c.1906/1907C>T和c.1971T>G基因突变患者表现为Rieger异常[2,3]。
INSR-IRS-1-PI3K-AKT信号通路通过调节外周胰岛素靶组织葡萄糖转运蛋白-4(Glucose transporter-4,GLUT-4)和GLUT-2的表达及转位介导葡萄糖摄取,该信号通路的任何基因突变均可导致胰岛素抵抗及糖尿病的发生。PIK3R1基因突变导致的PI3K调节亚基p85α功能缺失使其不能与催化亚基p110结合,致使INSR信号通路受阻导致胰岛素抵抗及糖尿病。本例患者表现为空腹正常或偏低血糖,餐后血糖异常升高,这种昼夜节律的血糖模式也见于INSR杂合突变导致的胰岛素抵抗患者,这是INSR及受体后信号通路障碍导致胰岛素抵抗患者的血糖谱特点,少食多餐或低碳水高蛋白质饮食可改善此种血糖的昼夜节律波动。该患者正常饮食或高碳水化合物饮食时血糖明显升高,但生酮饮食及禁食情况下血糖可维持在正常范围,符合PIK3R1基因突变致SHORT综合征糖尿病的血糖谱特点。二甲双胍是治疗SHORT综合征胰岛素抵抗及糖尿病的关键药物,但也有报道二甲双胍有加重胰岛素抵抗的可能。在二甲双胍单药治疗疗效差时,加用钠-葡萄糖协同转运蛋白2抑制剂(SGLT2i)也可获得良好疗效,SGLT2i不仅可通过降低肾糖阈、增加尿糖排泄量来降低日常食物摄入后导致的高血糖,而且还可以缓解轻微的夜间低血糖,但存在患者体重进一步减轻的风险[4,5]。本例患者使用二甲双胍治疗后空腹血糖达标但餐后血糖仍较高,结合患者高碳水化合物饮食后餐后血糖异常升高的血糖谱特点,同时避免使用SGLT2i使用后引起的体重进一步下降,给予患者二甲双胍联合阿卡波糖治疗,患者餐后血糖平稳达标。本例患者的治疗方案为SHORT综合征糖尿病的治疗提供了新方案。
SHORT综合征伴有身材矮小,男性成人身高为153.7~167cm,女性身高为141~167cm,生长激素治疗是增加终身高的有效方法,但会导致胰岛素抵抗的风险增加,尤其是合并糖尿病的患者会导致血糖难以控制,对于SHORT综合征患者,应谨慎评估生长激素治疗的益处和风险。本例患者已合并糖尿病,且骨骺已闭合,未再使用生长激素治疗。SHORT综合征亦会因胰岛素抵抗导致PCOS,本例患者虽妇科超声未达到PCOS诊断标准,但第二性征发育好,骨骺已闭合,至今无月经来潮,实验室检查提示睾酮及LH升高,仍考虑存在PCOS,给予二甲双胍治疗3个月后随诊,如仍无月经来潮可加用噻唑烷二酮类药物治疗。
参考文献:
[1] Ogawa W, Araki E, Ishigaki Y, et al. New classification and diagnostic criteria for insulin resistance syndrome.Endocr J. 2022;69(2):107-113.
[2] Shvalb NF. SHORT Syndrome: an Update on Pathogenesis and Clinical Spectrum.Curr Diab Rep. 2022;22(12):571-577.
[3] Innes AM, Neufeld S, Dyment DA.PIK3R1-Related SHORT Syndrome. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds.GeneReviews®. Seattle (WA): University of Washington, Seattle; May 15, 2014.
[4] Masunaga Y, Fujisawa Y, Muramatsu M, et al. Insulin resistant diabetes mellitus in SHORT syndrome: case report and literature review.Endocr J. 2021;68(1):111-117.
[5] Yang Y, Tan SHC, Lim SC, Loh WJ. Diabetes mellitus in SHORT syndrome managed with multi-agent oral therapies: a case report and literature review.Ther Adv Endocrinol Metab. 2025;16:20420188251405363. 2 comments










 京公网安备 11010502033361号
京公网安备 11010502033361号
发布留言