编者按:
胰高血糖素,这个与胰岛素“唱反调”的激素,传统上被视为维持血糖稳定的关键角色——空腹时促进肝糖原分解和糖异生,同时加速脂肪分解,为机体提供能量。然而,2型糖尿病(T2DM)患者长期处于病理性的高胰高血糖素状态。这会带来什么后果?
4月17日,在湘雅糖尿病免疫学大会(第二十一届湘雅论坛)基础与转化会场,中国医学科学院药物研究所李平平教授介绍了团队的最新研究进展:长期高浓度胰高血糖素,会使其生理作用发生根本性“反转”——从促进脂质分解转向促进脂质合成,进而直接驱动糖尿病肾病(DKD)的进展。该研究结果[1]已正式发表于权威期刊Nature Communications(自然 通讯)。
一、疑窦初现:高血糖之外,是什么推动了DKD?
DKD是糖尿病最严重的微血管并发症之一,临床表现为尿白蛋白/肌酐比值(UACR)≥30 mg/g和/或估算肾小球滤过率(eGFR)<60 mL/min/1.73 m2,病理特征包括肾小球硬化、肾小管肥大、基底膜增厚和间质纤维化。约40%的糖尿病患者会进展为DKD,是终末期肾病(ESRD)的主要原因。
高血糖是DKD的主要驱动因素。良好血糖控制可使1型糖尿病(T1DM)患者的DKD风险降低约1/2,T2DM患者降低约1/3。然而,这一数据也意味着:即使严格控制血糖,仍有相当比例的患者无法避免DKD的进展。这提示,除了高血糖,还存在其他促进DKD的关键因素。
T2DM患者普遍存在高胰高血糖素血症。临床数据显示,空腹胰高血糖素水平与UACR呈正相关,与eGFR呈负相关[2]。然而,这种高胰高血糖素血症是DKD的“结果”还是“原因”,目前尚不明确。这带来一个关键问题:高胰高血糖素血症是否直接介导并促进DKD进展?
二、层层剖析:胰高血糖素可直接促进DKD
为了回答这个问题,李平平教授团队设计了一套环环相扣的动物实验,从“相关”到“因果”,再到“反向验证”,层层解锁胰高血糖素与DKD之间的真实关系。
Step 1
明确关联——高胰高血糖素血症与DKD形影不离
团队首先在db/db小鼠(瘦素受体敲除DKD模型)及HFD+UniNx+STZ诱导的DKD小鼠中进行了系统检测。
结果发现,DKD小鼠体内呈现出三个关键特征:循环胰高血糖素水平显著上调(约2~4倍),胰高血糖素受体在DKD肾脏中表达显著增加,且该受体主要定位于肾脏皮质的近端肾小管上皮细胞。
这三条线索共同指向一个方向:高胰高血糖素血症与DKD之间存在紧密关联。但关联不等于因果,胰高血糖素是“旁观者”还是“肇事者”?需要进一步验证。
Step 2
建立因果——外源性胰高血糖素直接加剧DKD
为了从“相关”走向“因果”,团队直接给DKD早期小鼠注射外源性胰高血糖素,人为升高其胰高血糖素水平,观察DKD的进展。
结果令人警醒:UACR升高1.6倍,肾小管损伤标志物(Kim-1)升高1.5倍,肾脏重量增加1.25倍(图1)。
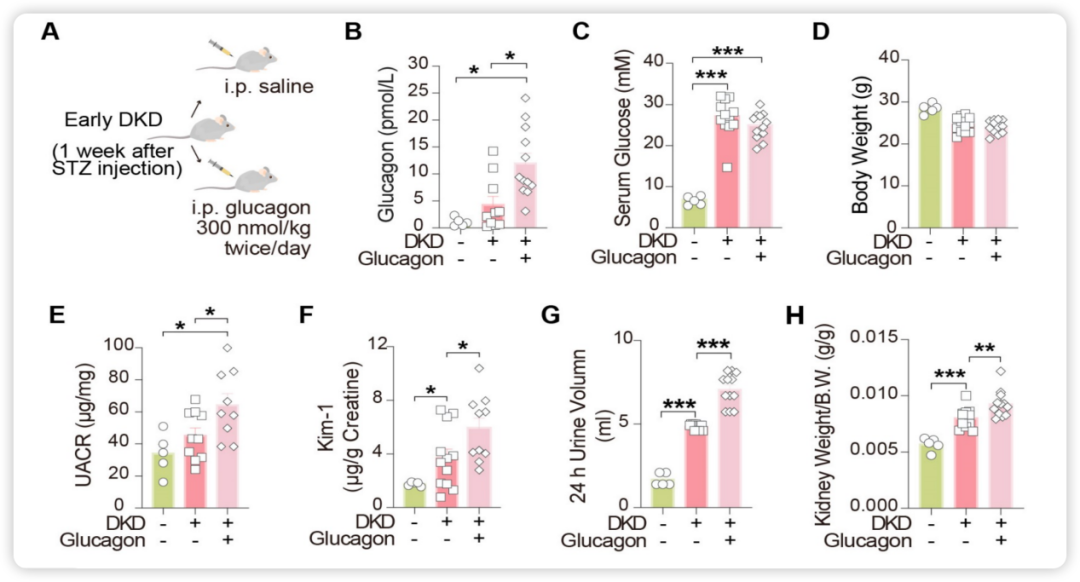
图1. 外源性胰高血糖素注射后,UACR、Kim-1及肾脏重量均显著升高
反向验证——阻断胰高血糖素受体“拯救”肾脏
因果关系的最高证据,不仅在于“给予”能加重疾病,更在于“阻断”能阻止疾病。
团队进一步采用基因敲除策略,特异性敲除肾小管上皮细胞的胰高血糖素受体。结果令人振奋:UACR近乎恢复正常,肾小管损伤标志物(Kim-1、NAG)显著下降,24小时尿量和肾脏重量恢复正常,脂质堆积和肾纤维化几乎完全消失,肾小球基底膜增厚和足细胞损伤也得到明显改善(图2)。阻断胰高血糖素信号,几乎完全阻止了DKD的进展。
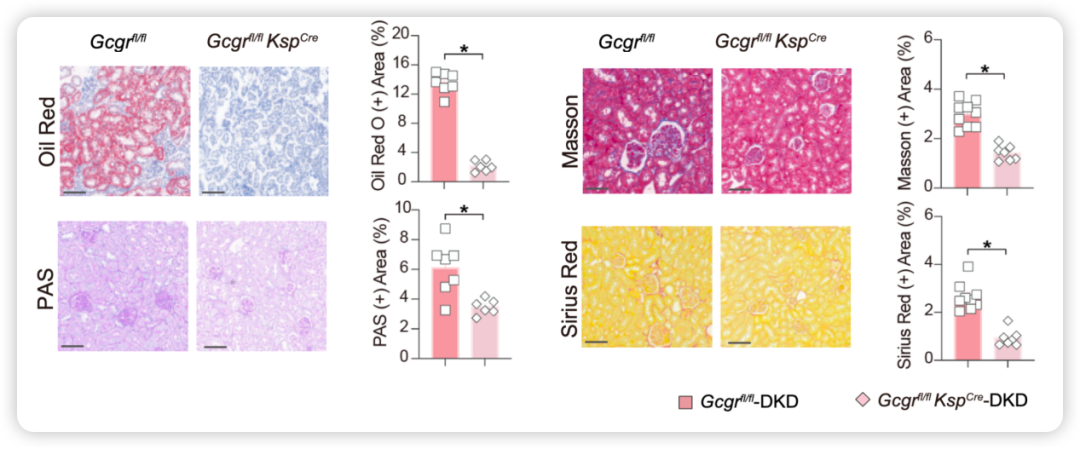
图2. 肾小管特异性敲除 Gcgr 改善 DKD 脂质堆积和纤维化
至此,三步实验环环相扣,形成了一个完整的证据链,无可辩驳地证明了:高胰高血糖素血症是DKD的独立驱动因素,而靶向其受体,则可能是治疗DKD的全新策略。
三、刨根究底,从“脂质分解”到“脂质合成”的反转
在明确了胰高血糖素可直接促进DKD后,团队对可能的致病机制进行了深入研究。
Step 1
现象——脂质堆积是DKD的“罪证”
正常情况下,肾脏中脂肪酸的摄取、氧化和合成处于精密平衡状态,不发生脂质堆积。然而,在DKD条件下,这一平衡被打破,脂质堆积已被证实是早期DKD进展的重要标志[3]。在DKD患者和动物模型中,肾脏脂质积聚与肾功能下降密切相关。
这不禁让研究者追问:是什么打破了这一平衡?
Step 2
反转——胰高血糖素长期暴露的“变脸”
带着上述疑问,团队对胰高血糖素的作用进行了时间梯度实验,结果发现了一个颠覆性的现象:
短期暴露(30分钟至4小时)时,胰高血糖素仍发挥其经典生理作用——促进脂质分解、增加能量产生;
然而,当暴露时间延长至24~48小时,胰高血糖素的作用竟然发生了根本性“反转”——从促进脂质分解转向促进脂质合成,能量产生也显著下降。长期高浓度胰高血糖素使脂质合成基因(FASN、SCD1等)表达显著上调,脂质转运和堆积增加,而脂质分解基因表达下调。
这一胰高血糖素作用反转的发现,用中国古话概括,正是“过犹不及,物极必反”。
Step 3
通路——GCGR-cAMP-PKA-CREB-RRAGA/B-mTORC1的完整链条
作用反转的现象固然惊人,但其背后的分子机制是什么?
团队进一步研究发现,长期高浓度胰高血糖素并不抑制胰高血糖素受体(GCGR)信号传导,反而通过cAMP-PKA-CREB通路,直接上调RRAGA/B的表达,进而激活mTORC1——一个经典的促进合成代谢(蛋白质、脂质、核苷酸)的分子开关(图3)。激活的mTORC1反过来促进脂质合成基因表达,最终导致脂质堆积、肾小管损伤和DKD进展。这一完整的信号链条可概括为:
高浓度胰高血糖素 → 激活GCGR → cAMP-PKA-CREB → 上调RRAGA/B → 激活mTORC1 → 促进脂质合成 → 脂质堆积 → 肾小管损伤 → DKD进展
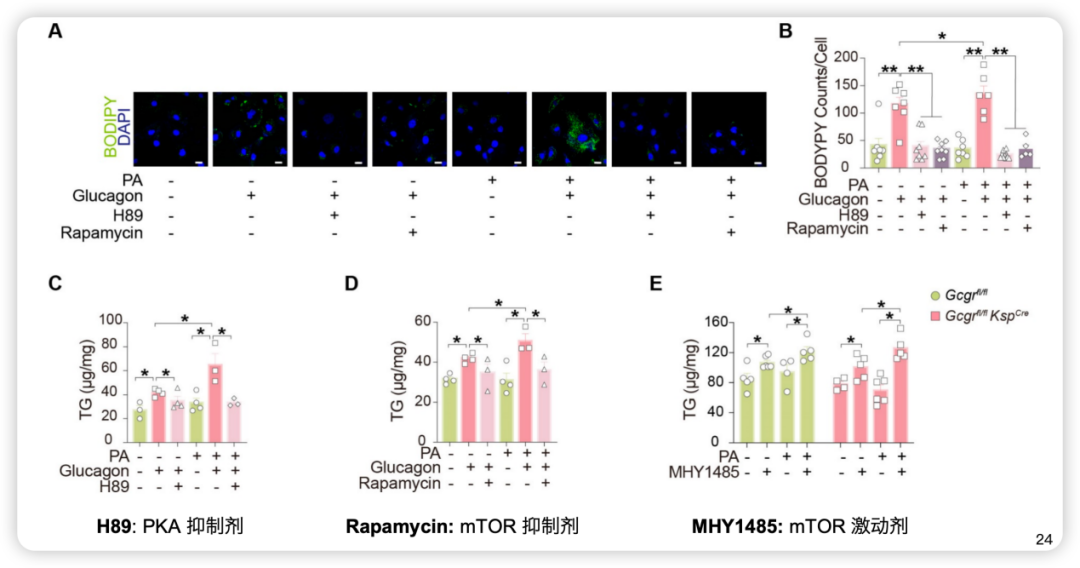
图3. 胰高血糖素促进脂质合成依赖PKA和mTORC1
关键实验证据为这一链条提供了有力支撑:使用PKA抑制剂(H89)或mTOR抑制剂(雷帕霉素)可完全阻断胰高血糖素诱导的脂质合成;而mTOR激动剂(MHY1485)则可模拟胰高血糖素的脂质堆积效应。
至此,从现象到机制,从宏观到微观,胰高血糖素“变脸”驱动DKD的完整故事被清晰呈现。
四、转化价值:GCGR抗体治疗DKD
研究进行到这里,一个关键问题自然浮现:既然胰高血糖素通过GCGR-mTORC1通路驱动DKD,那么阻断这条通路,能否逆转或阻止疾病进展?团队以此为出发点,将基础发现转化为治疗探索。
从实验室到动物模型:
GCGR抗体的初试锋芒
团队首先使用GCGR单克隆抗体(REMD 2.59)对2型糖尿病DKD小鼠进行了治疗。结果显示,抗体治疗组的UACR显著下降,肾小管损伤标志物明显改善,血清和肾脏中的脂质堆积减少,肾脏微结构病变得到缓解,异常激活的mTORC1信号也被有效抑制。
更值得一提的是,GCGR抗体的肾脏保护作用并不局限于2型糖尿病相关的DKD。在1型糖尿病引起的DKD模型中,即便GCGR抗体未能降低高血糖水平,仍可显著遏制DKD的进展(图4)。该结果证实:GCGR介导的肾损伤机制独立于高血糖之外。这也恰好解答了前文提及的临床长久困惑——为什么部分患者血糖已经控制得很好,最后仍会出现DKD进展。
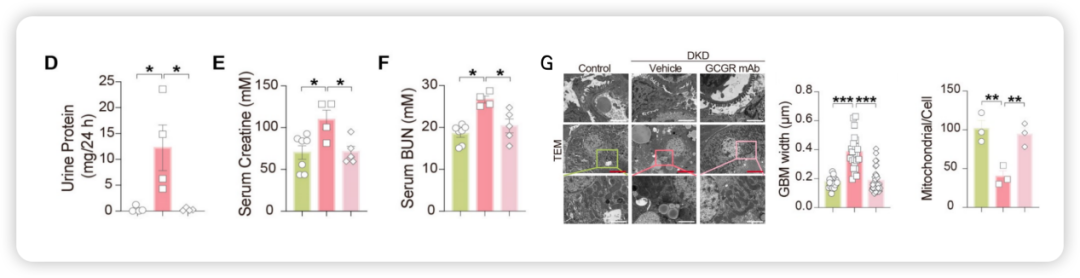
图4. GCGR抗体改善1型糖尿病引起的DKD
这一发现意义重大——它意味着胰高血糖素/GCGR信号通路不是某一类糖尿病的特例,而是不同类型DKD中普遍存在的致病机制。换句话说,无论是哪一类型的糖尿病,高胰高血糖素血症都在悄悄“助攻”肾脏损伤,而阻断这条通路,则有望成为共同的治疗策略。
至此,整个研究从一个看似矛盾的临床观察,揭开一个全新的病理机制,开辟一个潜在的治疗方向——这正是科学研究的魅力所在。
结语
从临床观察中的矛盾现象出发,到动物实验的层层验证,再到分子机制的深度解析,最后回归治疗探索——这项研究完成了一个完整的科学闭环。它揭示了一个被长期忽视的事实:在糖尿病长期高胰高血糖素血症的病理环境下,胰高血糖素会发生“角色反转”,从生理状态下促进脂质分解的“好帮手”,变成病理状态下促进脂质堆积、驱动肾损伤的“助推器”。
这一“物极必反”的发现,不仅改写了我们对胰高血糖素生物学效应的认知,更为DKD的治疗开辟了全新的靶点方向。GCGR抗体的成功应用,为那些即使严格控制血糖仍无法避免肾损伤的患者带来了新的希望。
专家简介

李平平
博士 研究员
中国医学科学院药物研究所博士生导师。在美国加州大学圣迭戈分校先后做博士后和助理教授。致力于肥胖及相关代谢疾病发病新机制、治疗新靶点和原创新药研究。以通讯(含共同)在 Cell、Nature Medicine、Nature Communications、Hepatology、APSB 等期刊发表数篇论文。先后获得国家高层次人才青年项目、国家优秀青年科学基金和国家青年科学基金项目(A类)支持和北京卓越青年科学家、树兰医学青年奖、求是杰出青年科学家和中国青年科技奖称号。
参考文献
[1] Liu X, Chen J, Gu S, et al. Nat Commun. 2025;16:8561.
[2] Li XC, et al. Clin Sci (Lond). 2008;114(9):591-601.
[3] Mitrofanova A, et al. Nat Rev Nephrol. 2023;19(10):629-645. 2 comments
胰高血糖素,这个与胰岛素“唱反调”的激素,传统上被视为维持血糖稳定的关键角色——空腹时促进肝糖原分解和糖异生,同时加速脂肪分解,为机体提供能量。然而,2型糖尿病(T2DM)患者长期处于病理性的高胰高血糖素状态。这会带来什么后果?
4月17日,在湘雅糖尿病免疫学大会(第二十一届湘雅论坛)基础与转化会场,中国医学科学院药物研究所李平平教授介绍了团队的最新研究进展:长期高浓度胰高血糖素,会使其生理作用发生根本性“反转”——从促进脂质分解转向促进脂质合成,进而直接驱动糖尿病肾病(DKD)的进展。该研究结果[1]已正式发表于权威期刊Nature Communications(自然 通讯)。

一、疑窦初现:高血糖之外,是什么推动了DKD?
DKD是糖尿病最严重的微血管并发症之一,临床表现为尿白蛋白/肌酐比值(UACR)≥30 mg/g和/或估算肾小球滤过率(eGFR)<60 mL/min/1.73 m2,病理特征包括肾小球硬化、肾小管肥大、基底膜增厚和间质纤维化。约40%的糖尿病患者会进展为DKD,是终末期肾病(ESRD)的主要原因。
高血糖是DKD的主要驱动因素。良好血糖控制可使1型糖尿病(T1DM)患者的DKD风险降低约1/2,T2DM患者降低约1/3。然而,这一数据也意味着:即使严格控制血糖,仍有相当比例的患者无法避免DKD的进展。这提示,除了高血糖,还存在其他促进DKD的关键因素。
T2DM患者普遍存在高胰高血糖素血症。临床数据显示,空腹胰高血糖素水平与UACR呈正相关,与eGFR呈负相关[2]。然而,这种高胰高血糖素血症是DKD的“结果”还是“原因”,目前尚不明确。这带来一个关键问题:高胰高血糖素血症是否直接介导并促进DKD进展?
二、层层剖析:胰高血糖素可直接促进DKD
为了回答这个问题,李平平教授团队设计了一套环环相扣的动物实验,从“相关”到“因果”,再到“反向验证”,层层解锁胰高血糖素与DKD之间的真实关系。
Step 1
明确关联——高胰高血糖素血症与DKD形影不离
团队首先在db/db小鼠(瘦素受体敲除DKD模型)及HFD+UniNx+STZ诱导的DKD小鼠中进行了系统检测。
结果发现,DKD小鼠体内呈现出三个关键特征:循环胰高血糖素水平显著上调(约2~4倍),胰高血糖素受体在DKD肾脏中表达显著增加,且该受体主要定位于肾脏皮质的近端肾小管上皮细胞。
这三条线索共同指向一个方向:高胰高血糖素血症与DKD之间存在紧密关联。但关联不等于因果,胰高血糖素是“旁观者”还是“肇事者”?需要进一步验证。
Step 2
建立因果——外源性胰高血糖素直接加剧DKD
为了从“相关”走向“因果”,团队直接给DKD早期小鼠注射外源性胰高血糖素,人为升高其胰高血糖素水平,观察DKD的进展。
结果令人警醒:UACR升高1.6倍,肾小管损伤标志物(Kim-1)升高1.5倍,肾脏重量增加1.25倍(图1)。
图1. 外源性胰高血糖素注射后,UACR、Kim-1及肾脏重量均显著升高
这些数据清晰地表明:胰高血糖素确实可直接促进DKD进展。更为关键的是,在整个实验过程中,小鼠的血糖水平并未发生变化——这意味着胰高血糖素的致病作用是独立于高血糖的。
反向验证——阻断胰高血糖素受体“拯救”肾脏
因果关系的最高证据,不仅在于“给予”能加重疾病,更在于“阻断”能阻止疾病。
团队进一步采用基因敲除策略,特异性敲除肾小管上皮细胞的胰高血糖素受体。结果令人振奋:UACR近乎恢复正常,肾小管损伤标志物(Kim-1、NAG)显著下降,24小时尿量和肾脏重量恢复正常,脂质堆积和肾纤维化几乎完全消失,肾小球基底膜增厚和足细胞损伤也得到明显改善(图2)。阻断胰高血糖素信号,几乎完全阻止了DKD的进展。
图2. 肾小管特异性敲除 Gcgr 改善 DKD 脂质堆积和纤维化
至此,三步实验环环相扣,形成了一个完整的证据链,无可辩驳地证明了:高胰高血糖素血症是DKD的独立驱动因素,而靶向其受体,则可能是治疗DKD的全新策略。
三、刨根究底,从“脂质分解”到“脂质合成”的反转
在明确了胰高血糖素可直接促进DKD后,团队对可能的致病机制进行了深入研究。
Step 1
现象——脂质堆积是DKD的“罪证”
正常情况下,肾脏中脂肪酸的摄取、氧化和合成处于精密平衡状态,不发生脂质堆积。然而,在DKD条件下,这一平衡被打破,脂质堆积已被证实是早期DKD进展的重要标志[3]。在DKD患者和动物模型中,肾脏脂质积聚与肾功能下降密切相关。
这不禁让研究者追问:是什么打破了这一平衡?
Step 2
反转——胰高血糖素长期暴露的“变脸”
带着上述疑问,团队对胰高血糖素的作用进行了时间梯度实验,结果发现了一个颠覆性的现象:
短期暴露(30分钟至4小时)时,胰高血糖素仍发挥其经典生理作用——促进脂质分解、增加能量产生;
然而,当暴露时间延长至24~48小时,胰高血糖素的作用竟然发生了根本性“反转”——从促进脂质分解转向促进脂质合成,能量产生也显著下降。长期高浓度胰高血糖素使脂质合成基因(FASN、SCD1等)表达显著上调,脂质转运和堆积增加,而脂质分解基因表达下调。
这一胰高血糖素作用反转的发现,用中国古话概括,正是“过犹不及,物极必反”。
Step 3
通路——GCGR-cAMP-PKA-CREB-RRAGA/B-mTORC1的完整链条
作用反转的现象固然惊人,但其背后的分子机制是什么?
团队进一步研究发现,长期高浓度胰高血糖素并不抑制胰高血糖素受体(GCGR)信号传导,反而通过cAMP-PKA-CREB通路,直接上调RRAGA/B的表达,进而激活mTORC1——一个经典的促进合成代谢(蛋白质、脂质、核苷酸)的分子开关(图3)。激活的mTORC1反过来促进脂质合成基因表达,最终导致脂质堆积、肾小管损伤和DKD进展。这一完整的信号链条可概括为:
高浓度胰高血糖素 → 激活GCGR → cAMP-PKA-CREB → 上调RRAGA/B → 激活mTORC1 → 促进脂质合成 → 脂质堆积 → 肾小管损伤 → DKD进展
图3. 胰高血糖素促进脂质合成依赖PKA和mTORC1
关键实验证据为这一链条提供了有力支撑:使用PKA抑制剂(H89)或mTOR抑制剂(雷帕霉素)可完全阻断胰高血糖素诱导的脂质合成;而mTOR激动剂(MHY1485)则可模拟胰高血糖素的脂质堆积效应。
至此,从现象到机制,从宏观到微观,胰高血糖素“变脸”驱动DKD的完整故事被清晰呈现。
四、转化价值:GCGR抗体治疗DKD
研究进行到这里,一个关键问题自然浮现:既然胰高血糖素通过GCGR-mTORC1通路驱动DKD,那么阻断这条通路,能否逆转或阻止疾病进展?团队以此为出发点,将基础发现转化为治疗探索。
从实验室到动物模型:
GCGR抗体的初试锋芒
团队首先使用GCGR单克隆抗体(REMD 2.59)对2型糖尿病DKD小鼠进行了治疗。结果显示,抗体治疗组的UACR显著下降,肾小管损伤标志物明显改善,血清和肾脏中的脂质堆积减少,肾脏微结构病变得到缓解,异常激活的mTORC1信号也被有效抑制。
从2型到1型:
作用通用性的有力证明
更值得一提的是,GCGR抗体的肾脏保护作用并不局限于2型糖尿病相关的DKD。在1型糖尿病引起的DKD模型中,即便GCGR抗体未能降低高血糖水平,仍可显著遏制DKD的进展(图4)。该结果证实:GCGR介导的肾损伤机制独立于高血糖之外。这也恰好解答了前文提及的临床长久困惑——为什么部分患者血糖已经控制得很好,最后仍会出现DKD进展。
图4. GCGR抗体改善1型糖尿病引起的DKD
这一发现意义重大——它意味着胰高血糖素/GCGR信号通路不是某一类糖尿病的特例,而是不同类型DKD中普遍存在的致病机制。换句话说,无论是哪一类型的糖尿病,高胰高血糖素血症都在悄悄“助攻”肾脏损伤,而阻断这条通路,则有望成为共同的治疗策略。
至此,整个研究从一个看似矛盾的临床观察,揭开一个全新的病理机制,开辟一个潜在的治疗方向——这正是科学研究的魅力所在。
结语
从临床观察中的矛盾现象出发,到动物实验的层层验证,再到分子机制的深度解析,最后回归治疗探索——这项研究完成了一个完整的科学闭环。它揭示了一个被长期忽视的事实:在糖尿病长期高胰高血糖素血症的病理环境下,胰高血糖素会发生“角色反转”,从生理状态下促进脂质分解的“好帮手”,变成病理状态下促进脂质堆积、驱动肾损伤的“助推器”。
这一“物极必反”的发现,不仅改写了我们对胰高血糖素生物学效应的认知,更为DKD的治疗开辟了全新的靶点方向。GCGR抗体的成功应用,为那些即使严格控制血糖仍无法避免肾损伤的患者带来了新的希望。
专家简介
李平平
博士 研究员
中国医学科学院药物研究所博士生导师。在美国加州大学圣迭戈分校先后做博士后和助理教授。致力于肥胖及相关代谢疾病发病新机制、治疗新靶点和原创新药研究。以通讯(含共同)在 Cell、Nature Medicine、Nature Communications、Hepatology、APSB 等期刊发表数篇论文。先后获得国家高层次人才青年项目、国家优秀青年科学基金和国家青年科学基金项目(A类)支持和北京卓越青年科学家、树兰医学青年奖、求是杰出青年科学家和中国青年科技奖称号。
参考文献
[1] Liu X, Chen J, Gu S, et al. Nat Commun. 2025;16:8561.
[2] Li XC, et al. Clin Sci (Lond). 2008;114(9):591-601.
[3] Mitrofanova A, et al. Nat Rev Nephrol. 2023;19(10):629-645. 2 comments







 京公网安备 11010502033361号
京公网安备 11010502033361号
发布留言