编者按:2026年美国糖尿病协会科学年会(ADA2026)上,中国学者再添亮眼成果。天津医科大学总医院内分泌代谢科团队携两项基础研究亮相,分别从翻译组学新视角与关键分子调控机制层面取得重要突破。团队利用Ribo-seq技术首次绘制了急性高糖刺激下原代胰岛的全局翻译重编程图谱,并深入阐明了EMC2通过维持内质网转位通道功能调控胰岛素生物合成的关键机制。两项研究互为补充,为理解胰岛β细胞功能调控提供了全新线索。本刊特此报道,并邀请刘铭教授进行深度点评,以飨读者。
2685-P
王艺清,刘瑶,石春阳,刘铭,许晓希
天津医科大学总医院 内分泌代谢科
背景与目的
胰岛β细胞精准感知机体葡萄糖浓度变化,并快速启动胰岛素合成分泌程序,是维持血糖稳态的关键。既往研究证实,高糖可在转录水平上调控β细胞分泌通路、能量代谢及信号转导相关基因的表达,但介导急性葡萄糖刺激的即时翻译组动态调控机制尚不明确。传统多聚核糖体图谱技术是探究基因翻译调控的经典手段,但样本需求量大,难以适配稀缺的原代胰岛组织研究,且无法提供核糖体在mRNA上的精准定位并解析精细翻译调控模式。本研究创新性采用翻译组测序(Ribo-seq)技术,通过捕获核糖体保护的mRNA片段,实现核苷酸分辨率的核糖体定位检测,精准描绘急性高糖刺激下小鼠原代胰岛的全局翻译动态图谱。本研究系统阐释并量化高糖对各基因翻译效率的调控作用,深度挖掘传统测序技术无法识别的翻译动力学新型调控模式,从翻译调控新视角揭示胰岛β细胞应答葡萄糖刺激、调控胰岛素合成的分子机制。
方法
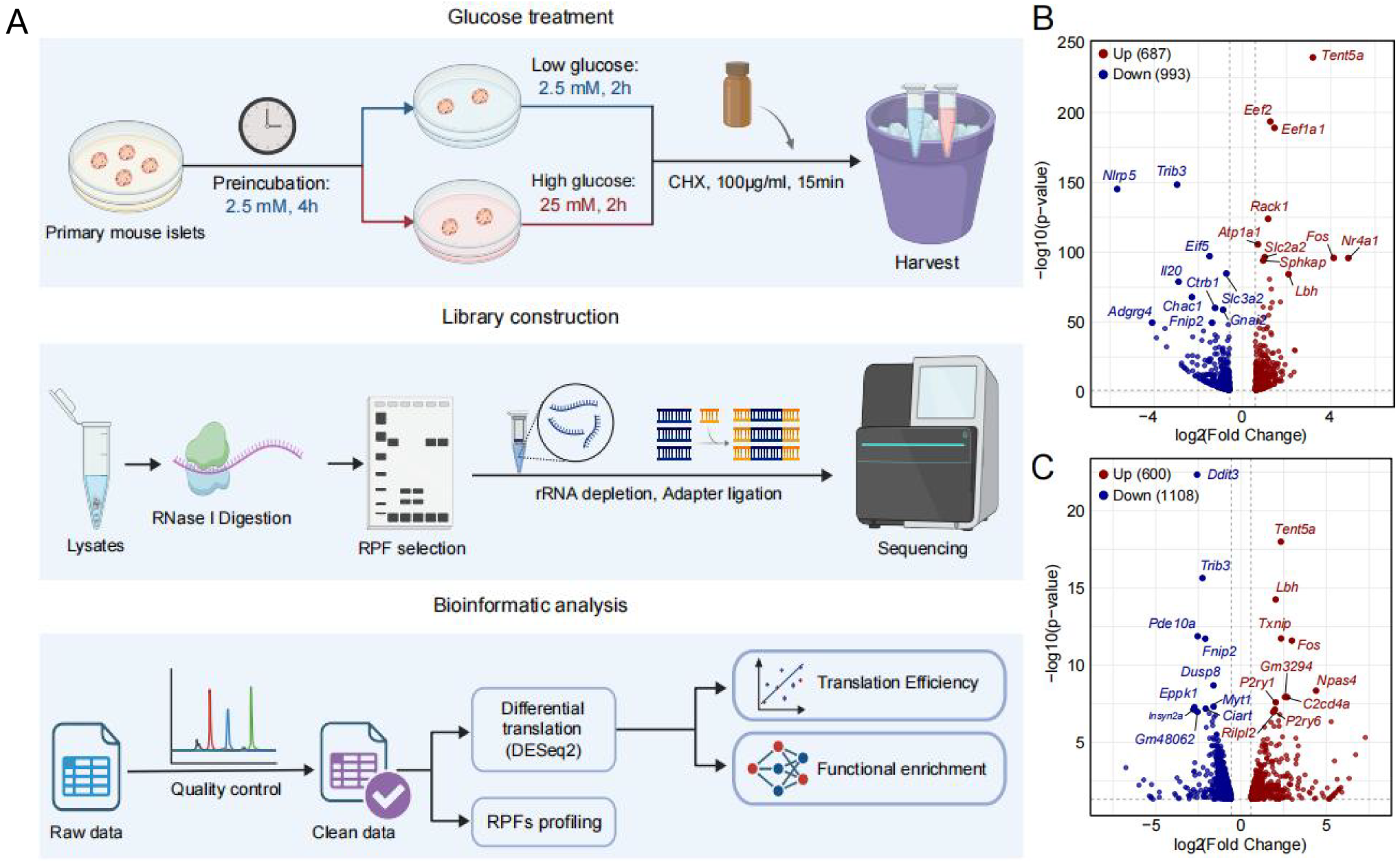
本研究选取6~8周龄C57BL/6J雄性小鼠,采用胶原酶消化法分离原代胰岛。无糖预处理4 h后,低糖组和高糖组分别置于2.5 mM与25 mM葡萄糖环境下继续培养2 h。样本收集前15 min,加入终浓度100 μg/ml的环己酰亚胺(CHX)固定核糖体,锁定瞬时翻译状态,随后收集胰岛样本并液氮速冻。研究采用Ribo-seq翻译组测序技术,系统筛选差异翻译基因,开展功能富集分析、翻译动力学解析、翻译效率定量(图1A)。同时结合蛋白免疫印迹(Western blot)、实时荧光定量PCR(qRT-PCR)实验,在分子水平验证关键基因的蛋白及mRNA表达差异。
结果
首先,急性高糖刺激可驱动小鼠原代胰岛发生广泛的翻译水平重塑,研究共筛选出1680个显著差异翻译基因,其中687个基因翻译上调(如即刻早期应答基因Fos、Nr4a1等)、993个基因下调(如应激凋亡相关基因Ddit3、Trib3等)(图1B和C)。
其次,急性高糖刺激可在翻译水平上呈剂量依赖性促进胰岛素前体翻译合成,而胰岛素mRNA表达无明显波动。翻译动力学分析进一步发现,高糖不仅能够改变核糖体整体密度,还可精准调控特定转录本的核糖体分布模式(图2),对Ins1等关键基因发挥特异性调控作用,但机制仍有待验证。
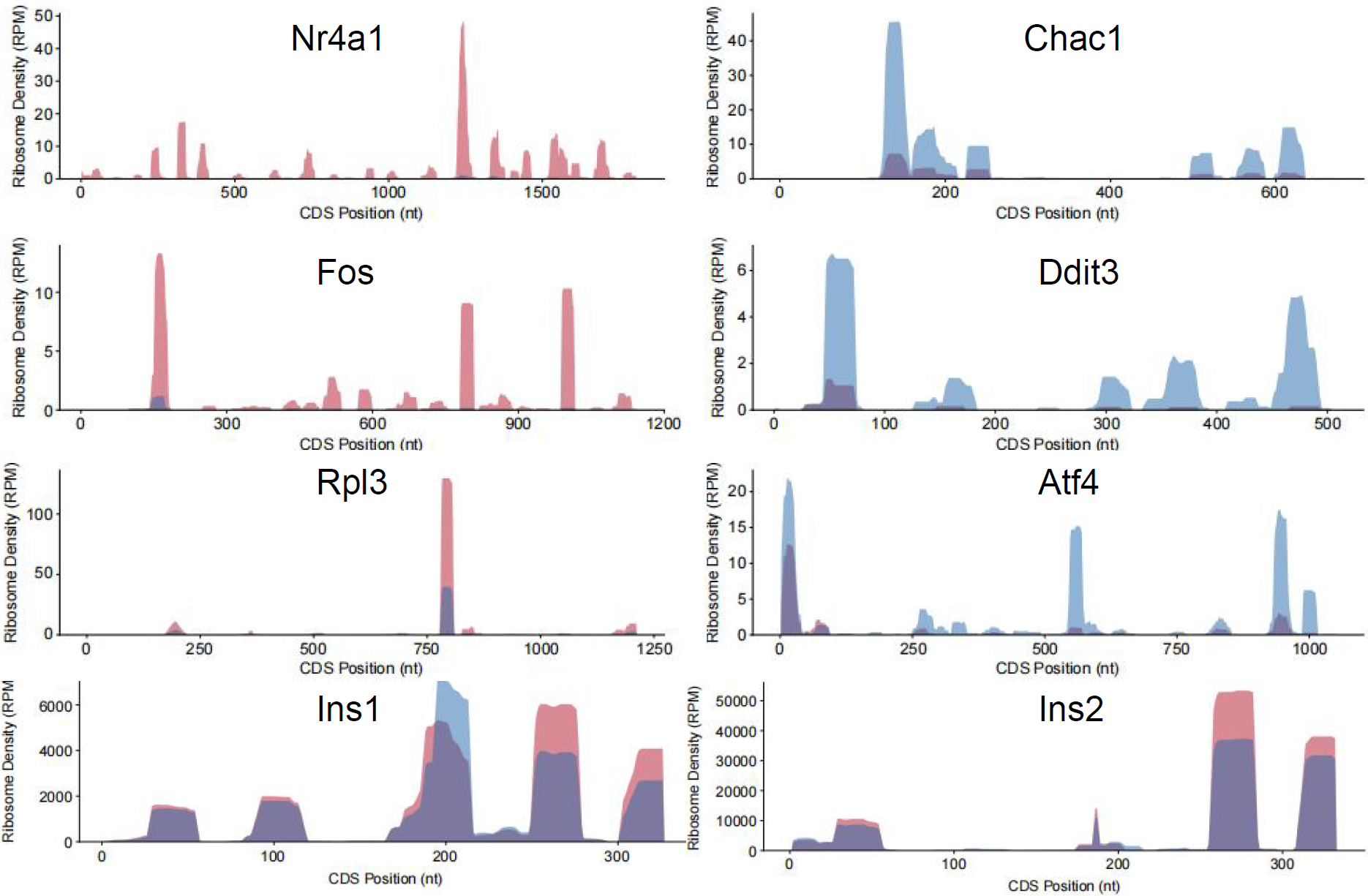
图2. 急性高糖刺激改变核糖体在特定转录本上的密度与分布
第三,高糖可在翻译水平协同重塑多条功能通路,优先上调Rpl3等胞质核糖体蛋白与翻译延伸因子,也促进胰岛素分泌途径相关基因(如内质网转位通道蛋白Sec61a1、囊泡转运组分Sec24a及胰岛素加工关键酶Pcsk1)的翻译。有趣的是,囊泡运输相关基因的翻译调控具有一定方向性,表现为胞吐相关基因翻译上调、胞吞相关基因翻译下调。
第四,高糖可在翻译水平精准重编程胰岛糖脂代谢基因表达。糖代谢通路中Slc2a2、G6pc2基因翻译上调最为显著;脂代谢通路呈现“合成激活、分解抑制”的调控特征。在线粒体代谢层面,高糖特异性上调编码三羧酸循环关键酶的基因翻译水平(Fh1、Cs等);但线粒体生物合成相关基因翻译广泛下调(Mrpls),与胞质核糖体的上调形成鲜明对比,呈现独特的代谢适配特征。
第五,mTORC1通路存在特殊的负反馈自限性调控机制。Western blot结果证实高糖可显著激活mTORC1通路,但通路核心组分编码基因的翻译水平反而下调(如Lamtor1等)。
最后,翻译效率定量分析证实,大量基因存在转录非依赖型翻译调控,形成多层次的翻译缓冲机制与细胞应激保护体系。
结论
本研究开创性建立了小鼠原代胰岛Ribo-seq翻译组测序标准化实验体系,首次系统展示急性葡萄糖刺激驱动原代胰岛产生全局性、协同性的翻译重编程效应。本研究从翻译调控的全新维度,筛选出一系列葡萄糖应答关键基因,为深入理解胰岛β细胞功能、阐明2型糖尿病(T2DM)发病的病理机制提供了翻译层面的参考,也为T2DM精准靶向治疗提供了全新的候选干预靶点。
EMC2调控胰岛β细胞胰岛素生物合成机制研究
2684-P
刘瑶,王艺清,冯文利,刘铭,许晓希
天津医科大学总医院内分泌代谢科
背景与目的
前胰岛素原经内质网Sec61转位通道跨膜转位,并加工成熟为胰岛素原,是胰岛素生物合成通路的首个限速环节,一旦发生缺陷会直接造成胰岛素合成受阻。但该过程潜在低效,高度依赖TRAP复合物、OST糖基化修饰复合物等多种蛋白机器辅助[1]。本课题组前期蛋白免疫共沉淀实验发现,Sec61转位通道蛋白的互作组分中显著富集内质网膜复合物(EMC)的多个亚基。EMC是真核生物内质网上高度保守的多功能膜蛋白复合物,由9个亚基组成,参与新生膜蛋白的内质网插入、膜折叠修饰与内质网稳态维持[2,3]。结构生物学研究发现,EMC2是维系EMC空间构象与功能完整性的核心亚基,是调控EMC功能的关键位点[4]。公共数据库数据分析显示,EMC2在T2DM患者胰岛组织中表达下调,提示其表达异常与胰岛β细胞功能衰退及T2DM发生发展密切相关,但机制尚不明确。基于此,本研究旨在探究EMC2对胰岛β细胞功能的调控,明确其在胰岛素生物合成中的作用。
方法
本研究以小鼠胰岛β细胞系Min6为研究模型,利用siRNA特异性敲低EMC2表达,同时设置转染scrambled siRNA的Min6细胞作为阴性对照。采用Western blot技术检测胰岛素及其前体、内质网转位通道核心组分的蛋白表达水平;通过ELISA实验检测EMC2缺失对胰岛素分泌功能的影响;运用SUnSET实验评估整体蛋白质合成能力的变化;RNA seq解析EMC2缺失诱导的细胞转录组紊乱特征,并通过qRT-PCR实验对关键差异基因的mRNA表达水平进行验证。
结果
研究表明,EMC2敲低的Min6细胞表现出胰岛素分泌能力受损,胞外胰岛素分泌量与胞内胰岛素储备的比值明显降低。EMC2敲降后,胰岛素前体——前胰岛素原及胰岛素原蛋白水平显著下降,但Ins1、Ins2胰岛素编码基因的转录水平无显著变化,提示EMC2缺失所致的胰岛素合成障碍发生于转录后水平。SUnSET实验结果进一步证实,EMC2敲低后嘌呤霉素掺入减少,胰岛β细胞整体蛋白质合成能力下降。此外,EMC2缺失下调内质网Sec61转位通道、TRAP复合物关键亚基的蛋白表达,破坏其结构完整性与功能稳定性。转录组测序分析表明,EMC2缺失可能诱发内质网稳态紊乱,进一步干扰胰岛素生物合成的下游通路。
结论
本研究发现,EMC2是维持胰岛β细胞中胰岛素生物合成的关键分子。其表达缺失会影响内质网转位通道功能,导致胰岛素原合成减少,并干扰内质网稳态,最终导致胰岛素合成与分泌功能双重缺陷。
专家点评

刘铭 教授
天津医科大学总医院
胰岛β细胞功能衰竭是糖尿病发生发展的核心因素,但深层次的调控机制仍未完全阐明。天津医科大学总医院内分泌代谢科团队在2026 ADA年会报道了两项基础研究。其中一项研究(2685-P)依托Ribo-seq翻译组测序技术,突破了传统转录组研究与多聚核糖体技术的局限性。该研究首次在小鼠原代胰岛中构建了急性葡萄糖刺激下的全局翻译重编程图谱,证实高糖可通过重塑全基因组翻译效率与翻译动力学,协同调控翻译机器、胰岛素生物合成、能量代谢、内质网转运及细胞稳态通路,为深入研究β细胞快速适配葡萄糖波动的核心分子机制提供重要参考,也为筛选糖尿病干预靶点提供了丰富的翻译组学资源。
另一项研究(2684-P)聚焦胰岛素生物合成的关键科学问题,基于前期互作组学基础展开探讨。研究发现,EMC2缺失导致胰岛β细胞全局蛋白合成能力下降、胰岛素前体合成减少,胰岛素分泌功能缺陷。EMC2作为EMC复合物核心骨架分子,可能通过维持内质网Sec61转位通道结构与功能完整,参与调控胰岛素生物合成。本研究为细胞实验,相关结果是否适用于原代胰岛可能还需进一步验证,具体分子机制也有待深入探讨。
参考文献:
1. Itskanov S, Park E. Mechanism of Protein Translocation by the Sec61 Translocon Complex. Cold Spring Harb Perspect Biol. 2023; 15(1): a041250.
2. Chitwood PJ, Hegde RS. The Role of EMC during Membrane Protein Biogenesis. Trends Cell Biol. 2019; 29(5): 371-384.
3. Wu H, Hegde RS. Mechanism of signal-anchor triage during early steps of membrane protein insertion. Mol Cell. 2023; 83(6): 961-973. e7.
4. Pleiner T, Tomaleri GP, Januszyk K, et al. Structural basis for membrane insertion by the human ER membrane protein complex. Science. 2020; 369(6502): 433-436. 2 comments

2685-P
王艺清,刘瑶,石春阳,刘铭,许晓希
天津医科大学总医院 内分泌代谢科
背景与目的
胰岛β细胞精准感知机体葡萄糖浓度变化,并快速启动胰岛素合成分泌程序,是维持血糖稳态的关键。既往研究证实,高糖可在转录水平上调控β细胞分泌通路、能量代谢及信号转导相关基因的表达,但介导急性葡萄糖刺激的即时翻译组动态调控机制尚不明确。传统多聚核糖体图谱技术是探究基因翻译调控的经典手段,但样本需求量大,难以适配稀缺的原代胰岛组织研究,且无法提供核糖体在mRNA上的精准定位并解析精细翻译调控模式。本研究创新性采用翻译组测序(Ribo-seq)技术,通过捕获核糖体保护的mRNA片段,实现核苷酸分辨率的核糖体定位检测,精准描绘急性高糖刺激下小鼠原代胰岛的全局翻译动态图谱。本研究系统阐释并量化高糖对各基因翻译效率的调控作用,深度挖掘传统测序技术无法识别的翻译动力学新型调控模式,从翻译调控新视角揭示胰岛β细胞应答葡萄糖刺激、调控胰岛素合成的分子机制。
方法
本研究选取6~8周龄C57BL/6J雄性小鼠,采用胶原酶消化法分离原代胰岛。无糖预处理4 h后,低糖组和高糖组分别置于2.5 mM与25 mM葡萄糖环境下继续培养2 h。样本收集前15 min,加入终浓度100 μg/ml的环己酰亚胺(CHX)固定核糖体,锁定瞬时翻译状态,随后收集胰岛样本并液氮速冻。研究采用Ribo-seq翻译组测序技术,系统筛选差异翻译基因,开展功能富集分析、翻译动力学解析、翻译效率定量(图1A)。同时结合蛋白免疫印迹(Western blot)、实时荧光定量PCR(qRT-PCR)实验,在分子水平验证关键基因的蛋白及mRNA表达差异。
结果
首先,急性高糖刺激可驱动小鼠原代胰岛发生广泛的翻译水平重塑,研究共筛选出1680个显著差异翻译基因,其中687个基因翻译上调(如即刻早期应答基因Fos、Nr4a1等)、993个基因下调(如应激凋亡相关基因Ddit3、Trib3等)(图1B和C)。
其次,急性高糖刺激可在翻译水平上呈剂量依赖性促进胰岛素前体翻译合成,而胰岛素mRNA表达无明显波动。翻译动力学分析进一步发现,高糖不仅能够改变核糖体整体密度,还可精准调控特定转录本的核糖体分布模式(图2),对Ins1等关键基因发挥特异性调控作用,但机制仍有待验证。
图2. 急性高糖刺激改变核糖体在特定转录本上的密度与分布
第三,高糖可在翻译水平协同重塑多条功能通路,优先上调Rpl3等胞质核糖体蛋白与翻译延伸因子,也促进胰岛素分泌途径相关基因(如内质网转位通道蛋白Sec61a1、囊泡转运组分Sec24a及胰岛素加工关键酶Pcsk1)的翻译。有趣的是,囊泡运输相关基因的翻译调控具有一定方向性,表现为胞吐相关基因翻译上调、胞吞相关基因翻译下调。
第四,高糖可在翻译水平精准重编程胰岛糖脂代谢基因表达。糖代谢通路中Slc2a2、G6pc2基因翻译上调最为显著;脂代谢通路呈现“合成激活、分解抑制”的调控特征。在线粒体代谢层面,高糖特异性上调编码三羧酸循环关键酶的基因翻译水平(Fh1、Cs等);但线粒体生物合成相关基因翻译广泛下调(Mrpls),与胞质核糖体的上调形成鲜明对比,呈现独特的代谢适配特征。
第五,mTORC1通路存在特殊的负反馈自限性调控机制。Western blot结果证实高糖可显著激活mTORC1通路,但通路核心组分编码基因的翻译水平反而下调(如Lamtor1等)。
最后,翻译效率定量分析证实,大量基因存在转录非依赖型翻译调控,形成多层次的翻译缓冲机制与细胞应激保护体系。
结论
本研究开创性建立了小鼠原代胰岛Ribo-seq翻译组测序标准化实验体系,首次系统展示急性葡萄糖刺激驱动原代胰岛产生全局性、协同性的翻译重编程效应。本研究从翻译调控的全新维度,筛选出一系列葡萄糖应答关键基因,为深入理解胰岛β细胞功能、阐明2型糖尿病(T2DM)发病的病理机制提供了翻译层面的参考,也为T2DM精准靶向治疗提供了全新的候选干预靶点。
EMC2调控胰岛β细胞胰岛素生物合成机制研究
2684-P
刘瑶,王艺清,冯文利,刘铭,许晓希
天津医科大学总医院内分泌代谢科
背景与目的
前胰岛素原经内质网Sec61转位通道跨膜转位,并加工成熟为胰岛素原,是胰岛素生物合成通路的首个限速环节,一旦发生缺陷会直接造成胰岛素合成受阻。但该过程潜在低效,高度依赖TRAP复合物、OST糖基化修饰复合物等多种蛋白机器辅助[1]。本课题组前期蛋白免疫共沉淀实验发现,Sec61转位通道蛋白的互作组分中显著富集内质网膜复合物(EMC)的多个亚基。EMC是真核生物内质网上高度保守的多功能膜蛋白复合物,由9个亚基组成,参与新生膜蛋白的内质网插入、膜折叠修饰与内质网稳态维持[2,3]。结构生物学研究发现,EMC2是维系EMC空间构象与功能完整性的核心亚基,是调控EMC功能的关键位点[4]。公共数据库数据分析显示,EMC2在T2DM患者胰岛组织中表达下调,提示其表达异常与胰岛β细胞功能衰退及T2DM发生发展密切相关,但机制尚不明确。基于此,本研究旨在探究EMC2对胰岛β细胞功能的调控,明确其在胰岛素生物合成中的作用。
方法
本研究以小鼠胰岛β细胞系Min6为研究模型,利用siRNA特异性敲低EMC2表达,同时设置转染scrambled siRNA的Min6细胞作为阴性对照。采用Western blot技术检测胰岛素及其前体、内质网转位通道核心组分的蛋白表达水平;通过ELISA实验检测EMC2缺失对胰岛素分泌功能的影响;运用SUnSET实验评估整体蛋白质合成能力的变化;RNA seq解析EMC2缺失诱导的细胞转录组紊乱特征,并通过qRT-PCR实验对关键差异基因的mRNA表达水平进行验证。
结果
研究表明,EMC2敲低的Min6细胞表现出胰岛素分泌能力受损,胞外胰岛素分泌量与胞内胰岛素储备的比值明显降低。EMC2敲降后,胰岛素前体——前胰岛素原及胰岛素原蛋白水平显著下降,但Ins1、Ins2胰岛素编码基因的转录水平无显著变化,提示EMC2缺失所致的胰岛素合成障碍发生于转录后水平。SUnSET实验结果进一步证实,EMC2敲低后嘌呤霉素掺入减少,胰岛β细胞整体蛋白质合成能力下降。此外,EMC2缺失下调内质网Sec61转位通道、TRAP复合物关键亚基的蛋白表达,破坏其结构完整性与功能稳定性。转录组测序分析表明,EMC2缺失可能诱发内质网稳态紊乱,进一步干扰胰岛素生物合成的下游通路。
结论
本研究发现,EMC2是维持胰岛β细胞中胰岛素生物合成的关键分子。其表达缺失会影响内质网转位通道功能,导致胰岛素原合成减少,并干扰内质网稳态,最终导致胰岛素合成与分泌功能双重缺陷。
专家点评
刘铭 教授
天津医科大学总医院
胰岛β细胞功能衰竭是糖尿病发生发展的核心因素,但深层次的调控机制仍未完全阐明。天津医科大学总医院内分泌代谢科团队在2026 ADA年会报道了两项基础研究。其中一项研究(2685-P)依托Ribo-seq翻译组测序技术,突破了传统转录组研究与多聚核糖体技术的局限性。该研究首次在小鼠原代胰岛中构建了急性葡萄糖刺激下的全局翻译重编程图谱,证实高糖可通过重塑全基因组翻译效率与翻译动力学,协同调控翻译机器、胰岛素生物合成、能量代谢、内质网转运及细胞稳态通路,为深入研究β细胞快速适配葡萄糖波动的核心分子机制提供重要参考,也为筛选糖尿病干预靶点提供了丰富的翻译组学资源。
另一项研究(2684-P)聚焦胰岛素生物合成的关键科学问题,基于前期互作组学基础展开探讨。研究发现,EMC2缺失导致胰岛β细胞全局蛋白合成能力下降、胰岛素前体合成减少,胰岛素分泌功能缺陷。EMC2作为EMC复合物核心骨架分子,可能通过维持内质网Sec61转位通道结构与功能完整,参与调控胰岛素生物合成。本研究为细胞实验,相关结果是否适用于原代胰岛可能还需进一步验证,具体分子机制也有待深入探讨。
参考文献:
1. Itskanov S, Park E. Mechanism of Protein Translocation by the Sec61 Translocon Complex. Cold Spring Harb Perspect Biol. 2023; 15(1): a041250.
2. Chitwood PJ, Hegde RS. The Role of EMC during Membrane Protein Biogenesis. Trends Cell Biol. 2019; 29(5): 371-384.
3. Wu H, Hegde RS. Mechanism of signal-anchor triage during early steps of membrane protein insertion. Mol Cell. 2023; 83(6): 961-973. e7.
4. Pleiner T, Tomaleri GP, Januszyk K, et al. Structural basis for membrane insertion by the human ER membrane protein complex. Science. 2020; 369(6502): 433-436. 2 comments









 京公网安备 11010502033361号
京公网安备 11010502033361号
发布留言